Lack of Evidence that Avian Oncogenic Viruses are Infectious for Humans: A Review
Published: May 14, 2015
By: Karel Schat AC and Hollis N. Erb B (A Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, NY; B Department of Population Medicine and Diagnostic Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY)
AVIAN DISEASES 58:345–358, 2014
Received 16 April 2014; Accepted 1 June 2014; Published ahead of print 1 June 2014
Received 16 April 2014; Accepted 1 June 2014; Published ahead of print 1 June 2014
Abstract
Chickens may be infected with three different oncogenic viruses: avian leukosis virus (ALV), reticuloendotheliosis virus (REV), and Marek’s disease herpesvirus (MDV). Several epidemiological studies have suggested a link between these viruses and different types of cancer in people working in poultry processing plants and with multiple sclerosis. In this article, we analyze the epidemiological evidence that these viruses are causative agents for human cancer, followed by description of the relevant key characteristics of ALV, REV, and MDV. Finally, we discuss the biological evidence or lack thereof that avian tumor viruses are involved in the etiology of human cancer and multiple sclerosis (MS). The recent primary epidemiologic articles that we reviewed as examples were only hypothesis-generating studies examining massive numbers of risk factors for associations with various imprecise, non-viral-specific outcomes. The studies lacked precise evidence of exposure to the relevant viruses and the statistical methods failed to adjust for the large risks of false-positive claims. ALV subgroups A–D and J have been eradicated in the United States from the pure lines down to the parent stocks by the breeder companies, which have greatly reduced the incidence of infection in layer flocks and broilers. As a consequence, potential exposure of humans to these viruses has greatly diminished. Infection of humans working in processing plants with ALV-A and ALV-B is unlikely, because broilers are generally resistant to infection with these two subgroups. Moreover, these viruses enter cells by specific receptors present on chicken, but not on mammalian, cells. Infection of mammalian cell cultures or animals with ALV-A, ALV-B, and ALV-J has not been reported. Moreover, humans vaccinated with exogenous or endogenous ALV-contaminated vaccines against yellow fever, measles, and mumps did not become antibody- or virus-positive for ALV. The risks for human infection with REV are similarly limited. First of all, REV also has been eradicated from pure lines down to parent stock by breeder companies in the United States. Broilers can still become infected with REV through infection with fowl pox virus containing REV. However, there is no indication that REV can infect human cells. Low levels of antibodies to ALV and REV in human sera have been reported by a few groups. Absorption of sera with chicken antigens reduced the antibody titers, and there was no clear association with contacts with poultry. Possible cross-reactions with human endogenous or exogenous retroviruses were not considered in these publications. MDV is typically associated with infection of chickens, and almost all experimental data show that MDV cannot infect mammalian cells or animals, including nonhuman primates. One study reports the presence of MDV gD DNA in human sera, but this finding could not be confirmed by another group. A Med-line search of the term ‘‘gene expression in human cancers’’ was negative for publications with avian retroviruses or MDV. In conclusion, there is no indication that avian oncogenic viruses are involved in human cancer or MS or even able to infect and replicate in humans.
Key words: avian leukosis virus, avian sarcoma virus, human cancer, hypothesis-generating study, Marek’s disease herpesvirus, multiple sclerosis, reticuloendotheliosis virus, Type-I error
Abbreviations: ALV=avian leukosis virus; CEF=chicken embryo fibroblasts; CI=confidence interval; COFAL=complement fixation for avian leukosis; DEF=duck embryo fibroblasts; EAV=endogenous avian retrovirus; EBV=Epstein-Barr virus; ev= endogenous viral; FFE=feather follicle epithelium; FPV=fowl pox virus; gs=group specific; HHV=human herpesvirus; HVT=herpesvirus of turkeys; ICD=International Classification of Diseases; IFAs=immunofluorescence assays; KSHV=Kaposi sarcoma-associated herpesvirus aka HHV-8; LTR=long terminal repeat; MDV=Marek’s disease virus; MS=multiple sclerosis; OR=odds ratio; PBL=peripheral blood lymphocytes; PMR=proportional mortality ratio; PPV=positive predictive value of the hypothesis test; REV=reticuloendotheliosis virus; RR=risk ratio; RSV=Rous sarcoma virus; SMR=standardized mortality ratio; SPF=specific pathogen free; SU=surface; TV=tumor virus; VN=virus neutralization; YF=yellow fever
Chickens harbor three groups of viruses that can cause tumors in chickens: avian leukosis viruses (ALV), reticuloendotheliosis virus (REV), and Marek’s disease virus (MDV). ALV and REV belong to the Retroviridae and MDV is a herpesvirus. Detailed reviews for ALV (98), REV (100), and MDV (126) have recently been published, and interested readers may want to consult these reviews for additional information. Concerns that these viruses may be able to cause disease in humans and therefore constitute a public health risk have been raised, especially since the first isolation of MDV in 1967 and the introduction of MDV vaccines shortly afterwards. Purchase and Witter (115) reviewed in 1986 the available evidence on Marek’s disease as a public-health risk factor and concluded ‘‘Thus in our opinion and in the opinion of experts in the human and avian cancer field, Marek’s disease and vaccines against Marek’s disease pose no human health hazards. Although additional studies are always possible, we believe there is sufficient evidence in this paper to warrant this firm conclusion.’’ In a recent review paper, Tischer and Osterrieder (140) also concluded that there is no formal proof or even likelihood of infection of humans with MDV. However, based on recent epidemiological studies, concerns have once more been raised that MDV and avian retroviruses can cause cancer in humans (33,48,69,72) and that MDV or the closely related herpes virus of turkeys (HVT), one of the MDV vaccine strains, may be the cause of multiple sclerosis (MS) (21,88,89,90,92,93). In this paper we will first analyze examples of the epidemiological evidence that these viruses are causative agents for human cancer, followed by description of the relevant key characteristics of ALV, REV, and MDV. Finally, we will discuss the biological evidence or lack thereof that avian tumor viruses are involved in the etiology of human cancer and MS.
EPIDEMIOLOGY REGARDING CANCER AND MORTALITY
Introduction.
Four recent primary epidemiological studies (46,48,68,71) have offered information on this topic; all are from the same research group and were made possible because of good access to union records for workers that included people in the livestock/abattoir industry in various states. There is also one recent review on this topic (69) from the same research group. The general aim of these papers was to investigate possible occupational relationships between exposure to livestock (especially poultry) and human cancers and mortality; each paper also included other risk factors not related to that general aim.
Study designs.
The four primary studies (46,48,68,71) were designed for hypothesis generation; none had an explicit refined primary hypothesis. Hypothesis generation is a useful goal when there is a lack of evidence to allow targeting of a very limited set of risk factors. Risk factors found to be significant in hypothesis-generating studies only provide ‘‘leads’’ to develop in more precisely focused hypothesis-testing studies. At some point, hypothesis-generating studies would be expected to lead to more explicit and limited hypotheses in, e.g., prospective cohort studies of workers in occupations speculated to be of increased risk. A cohort study design would allow direct focus on the risk factor of interest, because cohorts are groups of study subjects selected to be similar, except for the exposure to one risk factor designated a priori. Such prospective studies would necessarily be long term, but would have the opportunity to document exposures and specific disease outcomes better, including, perhaps, formal testing for the agents of interest.
All four primary studies used retrospective designs; that is, in each study the outcome (cancer or mortality) had already taken place when the research project was initiated. This is understandable and common for hypothesis-generating studies. Also, retrospective designs are especially efficient choices for outcomes such as cancer, in which the induction period is so long that many years have to pass before the outcome can be discerned credibly as present or absent. The disadvantage of retrospective studies is that the researcher either must use existing records or must interview people (and hope that memories are accurate).
Two of the primary studies compared the case group against the U.S. population (68,71). The other two studies used the case-cohort design. In the case-cohort design, cases are compared to a random sample of the people within the source population. Both designs are valid and are common in public health. In these four studies, the cases arouse from defined populations of ‘‘ever-workers’’ (e.g., members of the same union). Therefore, the two case-cohort studies might be slightly superior (compared to those of the other design) because the comparison group was also selected from ever-workers, and should have accounted for some general proxies for potential sources of confounding bias (see below).
The review article (69) is a systematic review that included only cohort studies; it explicitly excluded case-control studies. The systematic review does not adhere to the best reporting practices, as suggested by either Moher et al. (95) or Stroup et al. (139). For example, a few of the items missing from the report are the qualifications of the literature searchers, other fine details of the search strategy (e.g., keywords used and languages searched), how the authors searched for relevant unpublished studies, how abstracts and proceedings were used, how the risk and magnitude of reporting bias was assessed, and the table that describes each study included in the systematic review.
Johnson and Choi (69) chose not to run formal meta-analysis and gave several reasons. Some of the reasons imply that Johnson and Choi (69) do not accept the role meta-analysis can play in formally identifying and quantifying sources of heterogeneity or the manners in which alternative designs or studies of different perceived validity can be incorporated (20).
It is true that one should not try to combine alternative effect measurements (odds ratios [ORs], standardized mortality ratios [SMRs], risk ratios [RR], etc.) in a single meta-analysis; this was another reason given to avoid meta-analysis. Nevertheless, one can still run meta-analyses stratified on the more common effect measurements, and Johnson and Choi (69) included at least 42 studies in their review, so they could have run a meta-analysis on those. Also, one can sometimes go to the original papers and find enough data to allow calculation of alternative effect measures. No mention was made in Johnson and Choi (69) of whether this was attempted or how many studies might have had the same type of effect measure.
Finally, Johnson and Choi (69) indicated that some of the studies they found for the systematic review were of such poor quality that neither standard deviations nor confidence intervals (CIs) were presented—but it seems that those same articles were retained for the systematic review without clear designation in the text or tables as to which pieces of evidence were from the studies with the lower quality of statistical reporting.
Risk factors.
There was some general focus across the four studies on exposure to livestock (and in particular, poultry) or to their carcasses and products. This general focus explains the selection of many of the risk factors in each of the four example studies, and also explains the selection of some of the cohorts for which the records were reviewed. To their credit, those authors typically also tested many additional risk factors not related to livestock and poultry. Significance often was found for those other risk factors, as well as for some of the risk factors associated with livestock.
It is unlikely that interviews can be used to reconstruct much beyond ‘‘ever/never’’ responses for questions referring to several years past, and it appears that this is the type of question that typically was asked of respondents. What is lost, and typically was acknowledged by those authors, is any nuance regarding durations of exposures, doses of exposures, cumulative exposures, and timing of exposures relative to the outcome other than ‘‘before.’’ Such nuances would need to be studied with more-focused, prospective studies because such nuances probably are of interest, but this seems not to have been done.
A potential is present for information bias for the risk factors in Felini et al. (46) and in Gandhi et al. (48). The respondents for the risk factors for the cases were of necessity only next of kin (the cases were already dead). For the comparison cohort, the respondents were a mix of the subjects if alive and next of kin if the subject was already dead. Felini et al. (46) conducted a small (seven-pair) analysis of the agreement between the responses of live controls and their next of kin. For 245 yes/no questions, there was 100% agreement for 44% of the questions; agreement was only 60% to 79% for 18% (,44 questions), and 8% (20 questions) had ,60% agreement. Because only next of kin responded for the cases, this lack of even 80% agreement for a quarter of the yes/no questions applies differentially to the control group. The authors are to be commended for doing that small analysis. However, the authors neglected to note (e.g., on the results tables) which ,64 yes/no questions were the ones with poorer response agreement—especially whenever that risk factor was considered significant. Such questions would offer poorer leads than the questions with 100% agreement.
There appears to be some selective reporting of the risk factors found to be significant. It is the title, abstract, and key words that are indexed for other interested parties to find. Hypothesis-generating studies cannot be expected to list every risk factor tested when hundreds of risk factors are tested—nor even every risk factor found to be significant when there were dozens of those. On the other hand, authors can still indicate the numbers of risk factors tested so that the, albeit partial, list of significant risk factors declared in the abstract is put into perspective; this was not done in any of the four primary studies reviewed. In addition, Gandhi et al. (48) found only 13 significant risk factors even at their unadjusted alpha of 5%, and listing all 13 might well have been possible. However, the abstract listed only 5 of the 13 (including the only one related to poultry) without warning of another 8 significant risk factors or that the questionnaire from which the risk factors were derived contained 646 questions.
Outcome measurements.
In a retrospective study, the determination of the outcome must come from records or stored specimens. These four primary studies used records. The World Health Organization’s International Classification of Diseases (ICD) is standard for coding the ‘‘cause of death’’ in human beings in the United States. Johnson (68) and Johnson and Ndetan (71) were careful to indicate the ICD codes used to identify the various outcomes. However, Felini et al. (46) failed to do so, and Gandhi et al. (48) specified nothing more than the very general ICD code for ‘‘brain tumor’’ (without regard to, e.g., histopathological type or location within the brain).
Even where the ICD codes are given, one should remember that many of these ‘‘cause-specific codes’’ indicate only such characteristics as anatomic location, presence/absence of malignancy, and (sometimes) a histopathological descriptive diagnosis. It is doubtful that the histological diagnosis is pathognomonic for a particular cause for any of these studies, especially for such crude outcomes as e.g., ‘‘brain cancer’’, ‘‘liver cancer,’’ ‘‘pancreatic cancer,’’ or the dozens of diagnoses in Johnson (68). Such ICD codes do not state or imply the particular causal factors that interacted to produce the transformation to malignancy. Johnson and Ndetan (71) used a few ICD codes that imply microorganisms as causes of noncancer deaths, but even some of those (e.g., ‘‘other bacterial diseases’’) are imprecise. There are ICD codes for retroviral and herpesviral causes; neither these codes nor those for any other viral disease were included by Johnson and Ndetan (71), except to the extent that viral diseases would contribute to ‘‘all-causes death.’’
Control of confounding.
Confounding is a form of bias in which the true relationship between two variables is ‘‘confused’’ (confounded) because of the relationships of both to a third variable. The confounder must not be an intermediate link in the causal chain between the two variables in question, but is otherwise related to or predictive of both variables. Uncontrolled confounding can exaggerate, lessen, make nonsignificant, or even reverse the apparent relationship between the two variables in question; the direction and magnitude of the bias is not easily anticipated.
In observational studies, the control of potential confounding bias is accomplished by using one or more of four methods: restriction in the eligibility criteria, matching (not used in these four primary studies), stratification of the analysis, and statistical control in models. Each of these methods requires that the confounder be suspected a priori and that it be measured with precision and accuracy. It is difficult to identify potential confounders in a hypothesis-generating study, because so very many associations are tested—and each association might have its own unique confounders. Additionally, confounders might not be measured well in retrospective studies because of the problems arising from reliance on records and memories.
Eligibility criteria can be used to restrict the study subjects to people who have similar measurements for a potential confounder. When all subjects are alike, that confounder cannot differ between the study groups and confounding bias therefore cannot arise. The choice to use other members of the same union in the case-cohort studies probably lessened any confounding that might otherwise have arisen from something similar to the ‘‘healthy-worker effect’’ (confounding related to being both employable and willing to work). Gandhi et al. (48) pointed out that the selection of comparison groups from among the same union probably also accounts for a general level of ‘‘socioeconomic status’’ and any confounding that might arise from substantially different socioeconomic status. Use of cohorts from the same calendar years and age ranges also avoided some general sources of confounding. The studies variously used other forms of restriction (e.g., children hired at age ,10 yr were ineligible in Johnson [68]), but most additional restrictions seemed to be based on missing records or lack of interview with the next of kin.
Sex and race were considered potential confounders in Johnson (68) and in Johnson and Ndetan (71). Most analyses were stratified simultaneously on both male versus female and on white versus nonwhite race and also with some of those groups combined in additional analyses. Sex and race might still have confounded the combined-groups analyses; therefore, it might be prudent to use only the sex- and race-stratified results as leads for designing hypothesistesting research in the future.
Two of the primary studies used statistical analysis to control a small number of potential confounders. Felini et al. (46) adjusted for tobacco smoking and ‘‘newly identified risk factors’’ (not otherwise specified; p. 3). Felini et al. decided that age, race, and sex could be dropped from the models because the inclusion versus exclusion of these potential confounders did not alter the effect estimates for the risk factors being tested. This was an acceptable model-reduction decision because it was not based on the significance of the confounder per se. Gandhi et al. (48) adjusted for three locations of union sites and for age. Age was dichotomized bluntly at 50 yr. This categorization into just two groups might have been done to meet a key assumption of both the logistic and Cox (time-to-event) models. This assumption requires that the OR or HR for any risk factor is roughly the same for every 1-unit increase in the value of the risk factor; this must be true across the risk factor’s entire observed range of values, so is automatically met when there is only a single 1-unit increase possible (as when the data have been divided into only two groups). Although the control of age-related confounding was perhaps a little imprecise, the dichotomization made the models more credibly meet the assumptions and probably made them easier to fit for the pilot-study analysis.
The risk of false-positive claims of significance.
Each interpretation of statistical significance faces a risk that the interpretation is a mistake; in this mistake, the interpretation of significance is a false positive (i.e., a false claim, because the null hypothesis was true) (132). The simple probability of this risk is the alpha error (also known as the Type-I error). For a single test interpreted at alpha 5 5%, the risk is 1-in-20 that the interpretation of significance (with its rejection of the null hypothesis) is a mistake, and that the null hypothesis was true.
When many statistical hypothesis tests are run within a single study, this may lead to a multiplicity problem (132). If multiple statistical-hypothesis tests are run, the alpha-error risk increases rapidly. Running 100 tests for questions for which the null hypotheses are correct would expect to ‘‘find’’ something on the order of five false-positive claims. Unfortunately, we can never know which claims in a study are the false ones. One does not know that the null hypothesis is wrong or one would not be testing. If 100 tests were run and 10 were found to have P < 0.05 (if that’s the alpha), which of the 10 might be the expected false claims and which findings are the ones that are ‘‘true’’ cannot be known.
Some common protections for the multiplicity problem are to run as few tests as possible, to lower the alpha used to judge significance for individual tests, and to be constantly aware of the a priori chance that the alternative hypothesis (the claim of an effect or association) is true (see below). In addition, readers should always be aware of the alpha-error risk of false claims and of the need to use background knowledge and common sense in the interpretation of statistical results (62,105,132).
For hypothesis-generating studies (e.g., 46,48,68,71), some of these protections are not realistic. The idea of such studies is to run many tests to uncover new leads; if the authors already had strong a priori evidence of an association, the authors wouldn’t have had to generate leads—the authors already would have the leads. Such authors might impose some kind of adjustment to the alpha, but this was not done in the four primary studies reviewed. For example, Johnson (68) presented ,360 CI tests, each based on a 95% CI (equivalent to an alpha of 5% for each). If every null hypothesis were true, one would still expect to observe 18 false-positive claims. The tables (68) indicate significance 159 times (159 CIs excluded the null value); which of those are the 18 false claims expected just based on the 5% alpha cannot be known. One protection is simply being cautious in our interpretation.
The problem of false claims, however, runs even deeper for hypothesis-generating studies. The actual probability that a positive claim is a false positive (PPV; the positive predictive value of the hypothesis test) is based on more than just the alpha error. The sensitivity of the test and the prior odds that the alternative hypothesis is true also play a role (62,105). The sensitivity of the test is the power to detect that the null hypothesis is not true when it is indeed not true.
For hypothesis-generating studies, the prior odds are low almost by definition; such studies are looking for leads. At best, there might be some general focus (e.g., contact with livestock or work in a poultry abattoir)—but when scores or hundreds of risk factors are tested, the prior odds of a true association with the outcome for any single risk factor must be low (see the discussion of ‘‘Corollary 3’’ in Ioannidis [62]). The low prior odds then greatly decrease the PPVs in the study; i.e., the false-claim risk is increased far beyond the simple calculation based on alpha error.
A counter to Ioannidis’ Corollary 3 (62) is his Corollary 2 (paraphrased): larger effect sizes are somewhat more likely to be true than smaller effect sizes. He suggested designing studies to discover, e.g., RRs of at least 3 to 20 rather than RRs closer to the null value of 1.0. One might make use of this corollary in interpreting hypothesisgenerating studies (especially ones that made no attempt to adjust the alpha risk) by considering that only larger effect sizes suggest leads for targeted research. For the Johnson (68) study, the effect estimates were rough analogs to RRs: SMRs and proportional mortality ratios (PMRs). Imposing a criterion of a three-fold association (i.e., looking for SMRs and PMRs ≥3.0 or ≤0.33) would lower the number of leads to <30 from 159.
This same problem is present in the other studies reviewed. To the credit of the authors of Gandhi et al. (48), the potential is acknowledged in the discussion. Nevertheless, Gandhi et al. (48) asked respondents 646 questions; even if only 500 or 600 (of the 646) were tested as potential risk factors, most of the 13 odds ratios reported to be significant at 5% could be false-positive claims. The exception would be ionizing radiation, which was acknowledged by those authors on p. 5 as ‘‘the only known established risk factor for brain tumors.’’
Colinearity among risk factors.
Risk factors don’t only have relationships with the outcomes; risk factors also have relationships with other risk factors. Colinearity refers to the strength of such relationships among risk factors. Of course, with hundreds of tested risk factors, the same problems of false claims of relationships would necessarily arise if one tried to investigate all possible colinearities in the data set.
On the other hand, once a hypothesis-generating study has identified multiple leads, it might be prudent to investigate whether there are colinearities among those leads. If strong colinearities can be identified, then that is fair warning that there are fewer independent, distinct leads than thought. A colinearity investigation might also help tease out the significant risk factors on which to focus. For example, if one risk factor is strongly colinear with multiple other risk factors, then perhaps research might focus on the one for simplification; that one can also serve as a proxy for the others. It is unfortunate that none of the papers reviewed presented any analysis of colinearity.
AVIAN ONCOGENIC VIRUS INFECTIONS IN CHICKENS
Introduction.
In this section we will briefly review the current knowledge of the three avian oncogenic viruses and infection of chickens with ALV, REV, and MDV. As indicated before, detailed reviews of these viruses have recently been published.
Avian leukosis viruses and associated avian viruses.
ALV belongs to the Retroviridae, subfamily Orthoretrovirinae; the different strains of ALV are currently classified as part of the Alpharetrovirus genus (2). These viruses, like all members of the Retroviridae, are characterized by the presence of the pol gene that codes for two proteins: reverse transcriptase and integrase. In addition to pol, the genome of Alpharetrovirus consists of gag/pro and env genes. The gag gene codes for the structural core proteins, also known as groupspecific (gs) antigens. The env gene codes for the transmembrane and surface (SU) glycoproteins. The genome is flanked by long terminal repeats (LTR), which are important for the control of virus replication and cellular onc genes (98).
The ALV group contains six subgroups that can be present in chickens. Subgroups A–D and J are exogenous viruses, and subgroup E is an endogenous virus that is transmitted through the germ line. The endogenous virus is mostly present as defective endogenous viral (ev) sequences, but ev2 can produce subgroup E ALV (111,133). At least 29 ev sequences have been identified in the chicken genome (98), many of which are defective and not expressed. The finding that ev genes are not essential in chickens (3) led to the subsequent finding that additional endogenous provirus-like elements, referred to as endogenous avian retrovirus (EAV)-0, are present in the chicken genome independently of the presence of ev sequences (23,40). These EAV-0 elements were detected using gag, pol, env, and LTR sequences.
The exogenous viruses are transmitted horizontally and congenitally, but not through the germ line. Congenital transmission or horizontal transmission at a very early age after hatching often results in the development of viremic, antibody-negative chickens with a high incidence of B lymphocyte-derived lymphoid leukosis in the case of ALV-A or ALV-B or myeloid leukosis in the case of ALV-J (98). The subgroups ALV-C and ALV-D are very infrequently reported in poultry (98) and are not relevant for this review.
ALV infects cells through interactions between the SU glycoprotein and specific receptors (11). Briefly, three genes have been identified in chickens that control entry of ALV into the cell. The tumor virus (TV) genes conferring susceptibility to infection with ALV-A and ALV-C are named TVA*S and TVC*S, and the presence of alleles TVA*R and TVC*R confer resistance to infection. Susceptibility to ALV-B and ALV-D is governed by TVS*S1 for subgroups B and D and the resistance by S3TVB*R. One poultry breeding company has used this information and selected the pure lines for the alleles conferring resistance (McKay, pers. comm., 2014).
In addition to selecting for resistance, the poultry breeding industry has also implemented a highly successful eradication program for ALV-A and ALV-B since the early 1980s. The key to the eradication program was the finding by Spencer et al. (138) that gs antigens could be detected in the egg albumen, thus indicating that the hen was virus positive. The subsequent development of ELISA tests to detect p27, one of the gs antigens, allowed large-scale testing of eggs from the pure genetic lines for the four exogenous subgroups (137). The use of PCR technology provided an additional tool to eliminate eggs from positive hens (112). Because congenital transmission is the major route of infection and ALV is rather labile outside the host, it became possible to disrupt the transmission cycle of ALV-A, ALV-B, ALV-C, and ALV-D. Since the early 1980s, the grandparent and parent layer-type stocks from the major poultry breeders are free of ALV in the United States when the chicks leave the hatchery (McKay, pers. comm., 2014; Ansah, pers. comm., 2014). Although it is always possible that the parent flocks become infected afterwards, this is unlikely based on the absence of hosts other than chickens and on the thermal lability of retroviruses in general (98). However, ALV-associated outbreaks of tumors are occasionally reported in commercial layer flocks (53,156). The source of infection is difficult to identify, but one of the possible sources are the fancy breeds of chickens found in backyard flocks, which may be positive for ALV (150).
In 1991 Payne et al. (110) isolated a new subgroup ALV, from meat-type birds, that was subsequently named ALV-J. This virus shares the gs antigens with ALV-A to ALV-E, but the gene coding for the envelope glycoprotein is clearly different, with only 40% identity to the corresponding sequences of the other ALV subgroups (6). A large part of the env gene has homology with the EAV-E51 sequence (7). The development of ALV-J specific primers combined with ELISA tests for gs antigen, classical virus isolation methods, and serological assays allowed the poultry breeders to develop approaches for the eradication of ALV-J (134). In the United States, broiler grandparent and parent stocks from the major breeders have been free of ALV-J at hatch since the mid to late 1990s (McKay, pers. comm., 2014; Rossi, pers. comm., 2014), resulting in a reduction of outbreaks in parent flocks. As a consequence, requests for ALV-J diagnosis by PCR, as well as the incidence of positive cases, have declined drastically in one diagnostic facility (Fig. 1) (85).
Infection with exogenous ALV results in the insertion of the viral genome in the infected cells. Oncogenic transformation of the infected cells depends on the activation of cellular onc genes by the viral LTR (82). In some instances, a cellular onc gene has been transduced by ALV resulting in a defective virus which needs a helper ALV to infect cells. There is one known exception of a replication-competent virus with an onc gene: Rous sarcoma virus (RSV), which contains the src onc gene (42). RSV and defective leukemia viruses are of no practical importance in the poultry industry.
Fig. 1. Detection of avian leukosis virus subgroup J (ALV-J) by polymerase chain reaction (PCR) in broiler samples submitted to the Alabama Diagnostic Laboratory from 1998 to 2013 (85).
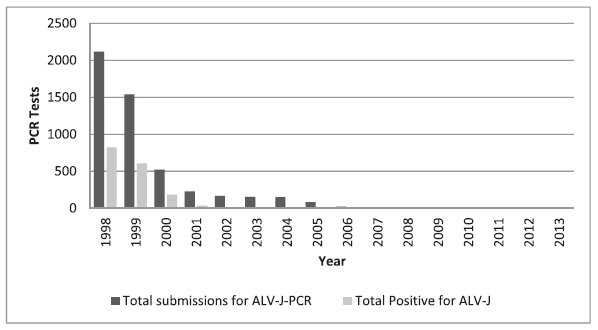
Based on the eradication of ALV subgroups from the pure lines to the parent stock since the early 1980s for layer-type chickens, the risk of human exposure to ALV-A and ALV-B has been greatly reduced, although not completely eliminated (98). Broilers, the main source of exposure to slaughterhouse workers, are considered more resistant to infection with ALV-A and ALV-B than layers (51) and there is very little information on the presence and shedding of ALV-A and ALV-B in broilers. ALV-J was a new virus first reported in 1991 (110) causing a rapid increase in infections in broilers but also a rapid reduction in apparent incidence after 2000 (Fig. 1).
Reticuloendotheliosis virus.
REV also belongs to the Retroviridae, subfamily Orthoretrovirinae, but is clearly different from the ALV group and is currently placed in the genus Gammaretrovirus (2).
The REV group is characterized by its close relationship to Echidna endogenous retrovirus and may have become adapted to birds through contaminated Plasmodium lophurae (malaria) stocks in ducks (104). In fact, REV shares some antigenic determinants with mammalian type C retroviruses (30) and specifically with MMC-1, a rhesus monkey endogenous type C retrovirus (108). There is no information on the possible presence of shared antigenic determinants with human endogenous viruses.
Transmission of REV in chickens can be horizontal or vertical, similar to the transmission of exogenous ALV (100). Horizontal transmission can be by direct contact, e.g., in the hatchery in the presence of vertical transmission (100), by insects (37), or by contaminated vaccines, especially fowl pox virus (FPV) vaccines. Hertig et al. (58) found that full-length, infectious REV genome can be integrated in the FPV genome. Many of the early FPV vaccines were contaminated with integrated REV, and that contributed importantly to the spread of REV in chicken populations, causing outbreaks of reticuloendotheliosis (49). Since 1999, FPV vaccines in the United Statesmust be free of REV (144) and the transmission of REV through vaccination with FPV vaccines no longer occurs. However, this is not necessarily the case in other parts of the world (87). Moreover, wildtype FPV may be positive for REV and infect chickens. MDV can also contain the LTR of REV (63,80), but REV sequences were not detected in the genomes of MDV vaccines (78,135,136).
Replication-competent REV causes tumors by insertion of the viral genome into the genome of infected cells and subsequent activation of cellular onc genes, especially c-myc (100). The defective strain T REV, containing the rel onc gene, can cause rapid development of tumors in chickens (100). The T strain needs a helper virus to infect cells and is not important for the poultry industry.
The breeder industry has used the same approach for the eradication of REV as for ALV. Pure lines, grandparent flocks, and parent flocks in the United States are free of REV at 1 day of age (McKay, pers. comm., 2014; Ansah, pers. comm., 2014). Infection in commercial chickens might still occur, but the frequency in broilers is probably low especially, because FPV vaccines have been free of REV since 1999 (144).
Marek’s disease virus and related herpesviruses.
MDV serotype 1 (MDV-1), also known as Gallid herpesvirus 2, belongs to the Mardivirus genus of the Alphaherpesvirinae subfamily of the Herpesviridae together with Gallid herpesvirus 2 (MDV serotype 2 [MDV-2]) and Meleagrid herpesvirus 1 (herpesvirus of turkeys [HVT]) (2). MDV-1 and MDV-2 are strictly cell associated in vivo except in the feather follicle epithelium (FFE) where cell-free virus is produced (27,125). Cell-free HVT is also produced in the FFE of turkeys and chickens (154) although horizontal transmission from chicken to chicken is limited at most (32). In contrast to ALV and REV, MDV is only horizontally transmitted; there is no egg transmission (126). Virus encased in keratin enters the bird through the respiratory tract, most likely by infecting macrophages, which transport the virus to the spleen. Based on splenectomy experiments, the spleen is probably the first lymphoid organ that becomes infected (123). Other lymphoid tissues such as the thymus and bursa of Fabricius also become infected very early after exposure. MDV-1 replication first occurs in the B lymphocytes and subsequently MDV-1 infects activated T lymphocytes, where it goes latent or causes transformation (26,126).
The mechanism of transformation has not completely been elucidated and several gene products play a role. A key protein for transformation is Meq, a bZIP protein, which can dimerize with itself or with the cellular oncoprotein Jun (99). Meq is expressed in transformed lymphocytes in tumors and cell lines which were developed from tumors (25,86,109,116,120) and is of diagnostic importance to identify MD tumor cells in chickens. Based on deletion mutants, additional gene products such as the viral RNA telomerase subunit (31,141) and viral interleukin-8 (66) also play a role in the transformation process. In contrast with MDV-1, MDV- 2 strains such as SB-1 and HVT are nononcogenic in chickens (125,151) even when inoculated in 8-day-old chicken embryos (29,125).
Since 1970, all commercial chickens in the United States have been vaccinated against MDV with HVT, HVT+SB-1 starting in 1983, or with CVI988, a MDV-1 strain (119) beginning in 1994. Broilers are generally vaccinated in ovo at 18 days of embryonation, whereas grandparent and parent flocks, are vaccinated in ovo and at 1 day of age; layer flocks are vaccinated at 1 day of age (24,52, Zavala, pers. comm., 2014). Vaccination not only reduces viremia, and disease after challenge (126), but also reduces shedding of the challenge virus (8,45). However, the absence of sterilizing immunity has been implicated in the evolution of MDV strains to increased virulence (4,5,152).
Importance of specific-pathogen-free chickens for vaccine production.
The realization that ALV and REV can be transmitted through embryonated chicken eggs used for human and animal vaccine production was a major impetus to develop specific-pathogenfree (SPF) flocks which became available in the United States in 1961 (Girshick, pers. comm., 2014). Currently in the United States, all chicken embryo-derived, live vaccines for humans (yellow fever [YF], measles, and mumps] are made in SPF substrates free from exogenous ALV and REV. However, these embryos may be positive for ev genes or EAV-0 sequences. TheWHOallows the production of YF vaccines in countries without SPF chicken flocks based on the low risks of ALV-contaminated vaccines for humans (54).
Although SPF-derived chicken embryo fibroblast cultures are used for MDV vaccines, occasionally serials can be found to be contaminated with ALV as described in References 44 and 155. This incident occurred with vaccine batches from two manufacturers containing different combinations of MDVvaccine strains, suggesting a common source of contamination. The cause of this occurrence has not been completely resolved; sequence information suggests a recombination of endogenous viral elements with exogenous ALV-A env sequences (10). The recombinant virus was growing slowly in cell culture (44,155). It was suggested that this, in combination with the extensive cytopathic effect of the vaccine viruses were the explanation that the complement-fixation for avian leukosis (COFAL) tests did not detect the presence of ALV in the vaccine batches. As a consequence, the COFAL test was replaced by the ELISA test for screening of vaccines for the presence of ALV (143), which is basically the same test commercial breeders have used since the early 1980s.
LACK OF EVIDENCE THAT AVIAN ONCOGENIC VIRUSES ARE INVOLVED IN HUMAN CANCER
Introduction.
As mentioned before, a series of recent epidemiology papers suggested that avian oncogenic viruses are associated with or are the cause of human tumors, although we believe this evidence to be tenuous for the reasons explained in the section ‘‘Epidemiology Regarding Cancer and Mortality.’’ Another suggestion of such an association arose because some researchers reported the presence of antibodies against ALV, REV, and MDV in human sera (35,73,74), and more recently, the presence of MDV sequences in human sera (83). However, it has to be realized that many human viruses are also associated with cancer: two human herpesviruses (Epstein-Barr virus [EBV]), Kaposi sarcoma-associated herpesvirus (KSHV/human herpesvirus [HHV]-8), several polyoma viruses, human papilloma viruses, hepatitis B virus, hepatitis C virus, and at least two but probably more human retroviruses (75). In the next sections we will analyze the available literature on the serology, virology, and pathology of avian oncogenic viruses in mammalian species, including human beings.
Serology.
The presence of antibodies against a pathogen does not necessarily indicate replication of the pathogen; it simply indicates exposure to one or more antigens associated with the pathogen. It is also important to realize that serological cross-reactions can occur. For example, cross-reactivity between pseudorabies virus (suid herpesvirus 1) and MDV has been described with the use of immunofluorescence assays (IFAs) with polyclonal (129) or monoclonal (103) antibodies.
Avian leukosis virus and reticuloendotheliosis virus.
In the past, human vaccines (YF, measles, and mumps) were prepared in embryonated chicken eggs or chicken embryo fibroblast (CEF) cultures known to be positive for exogenous ALV, which raised important questions on risks or evidence of exposure to ALV. After the establishment of SPF chicken flocks, vaccines were free from exogenous ALV, but not from endogenous ALV (subgroup E), ev gene products, or EAV-0 sequences. Reverse transcriptase activity associated with EAV-0 sequences was detected in vaccines derived from CEF or chicken embryos with the use of highly sensitive fluorescent product-enhanced reverse transcriptase assays (17,19,149). Several groups have examined human sera for the presence of antibodies to exogenous ALV after vaccination with ALV-contaminated YF, measles, and mumps vaccines. No matter which test was employed (COFAL, agar-gel precipitation, virus neutralization [VN]), the results were negative (55,91,113,118). A search for ALV antibodies in 103 sera from patients with different diagnoses of leukemia was also negative (121). Sera were also negative for ALV antibodies in Western blot assays after vaccination with vaccines containing endogenous ALV or EAV-0 sequences (60,61,142).
Choudat et al. (35) reported the presence of ALV antibodies in human sera using an in-house ELISA but there was no difference between groups exposed and unexposed to poultry. Sera were not absorbed with chicken powder prior to testing. On the other hand, using a commercial ELISA kit adapted for human sera, Johnson et al. (73) reported the presence of antibodies to ALV and REV in 19/ 45 and 9/45 samples, respectively. Sera were considered positive if the optical density was higher than the highest reading for the control sera. This approach has a problem because it is not clear if these ‘‘negative’’ sera are indeed negative (35) or in the ‘‘opposite’’ direction that the observed control titers are the highest that a negative subject can have. Moreover a standard for deciding positivity in an ELISA is frequently based on a value above the mean +2 or even 3 standard deviations of the controls (64). Absorption of the sera with CEF reduced the titer of the positive sera drastically; thus, a major part of the response was directed against chicken antigens. A small subset of reactive sera tested by Witter was negative for VN antibodies (73). In a second paper, Johnson et al. (74) used Western blot assays to analyze the positive responders in more detail. The highest ALV responder in the ELISA was only positive for p15, the virus-encoded protease. A basic local alignment search tool (BLAST) search was conducted indicating that there are several putative conserved domains between the ALV p15 amino acid sequence and human endogenous and exogenous retrovirus sequences. As a consequence, it cannot be excluded that the results reported by Johnson et al. (74) reflect the presence of antibodies to human endogenous or exogenous retroviruses. Results for env proteins were not available due to lack of purified env proteins. The summarized results for p27, p19, p15, and p12 of ALV and for REV did not show significant differences between poultry workers and controls. Moreover, REV shares antigenic determinants with p30 of mammalian retroviruses (9). Based on the ELISA data, the same group (33,34) reanalyzed the serological data in 2011 for links to age, job description, etc., and suggested that there were increased risks for ALV and REV in certain jobs compared to other jobs. However, the number of positive sera was rather limited, especially for REV, which prevents drawing meaningful conclusions. Moreover, the authors did not discuss in their analysis the fact that absorption with CEF reduced the responses nor that Western blot analysis did not show significant differences between poultry workers and controls.
Marek’s disease virus.
Antibodies in human sera to MDV were reported in the early 1970s by Japanese workers (76,101,102,107). However, they also reported cross-reactivity between EBV and MDV when using indirect immunofluorescence and agar gel precipitation assays. Sharma et al. (130) examined 205 human sera for antibodies to MDV and HVT by IFAs and found antibodies to MDV in 6.3% and to HVT in 2.9% of the sera. None of the positive sera had virus-neutralizing antibodies. Interestingly, the sera from 23 employees at a chicken processing plant were negative, whereas 4/50 sera from students without poultry contact were positive. In addition, 31 people with a known history of repeated accidental injections with HVT-contaminated needles did not develop antibodies to HVT, although 4 had low titers to MDV. Choudat et al. (35) used an ELISA developed in-house to examine the presence of MDV antibodies in sera from workers in three broad categories: poultry industry-associated, limited exposure to poultry, and without known contact with poultry. The OR to detect MDV antibodies was significantly higher in the exposed than in the unexposed groups (OR 6.17; 95% confidence interval 3.91–9.75). In addition, the titers were higher in the exposed groups. However, adsorption of sera with chicken powder reduced the optical densities of the ELISA readings and the only significant difference that remained was between exposed and unexposed women. This reduction in significance raises important questions concerning the relevance of this study as an indication of exposure to MDV.
Infection of mammalian cell cultures.
Avian retroviruses.
Exogenous ALV subgroups infect chicken cells by the interaction between the gp85 env protein and specific receptors, which are present in chickens but not in mammalian species. As a consequence subgroups A and B can infect chicken but not mammalian cells by binding to specific receptors as described in the section ‘‘Avian leukosis virus and associated avian viruses.’’ Duff and Vogt (39) reported in 1969 the presence of ‘‘contaminants’’ in subgroup A and B virus stocks. These viruses, identified as subgroup C and D, can infect mammalian cells (18,22,39). This finding explained also why previous work had suggested that mammalian species are susceptible to ALV-A and ALV-B. Boettiger et al. (18) noted that subgroup D viruses were all derived from strains that were adapted to mammalian cells or passed through mammalian hosts and that subgroup D viruses had not been isolated from the field.
In addition to the many avian cell types, REV can infect the canine D17 osteosarcoma cell line (9,147) and normal rat cells (77). Infection of human cells has been controversial. Koo et al. (79) showed that spleen necrosis virus (SNV), one of the REV viruses, could infect HeLa and COS-7 cells, but Gautier et al. (50) concluded that REV strains are not able to infect human cells. Both studies were done with replication-incompetent vectors expressing the REV env gene and expressing reporter genes.
Marek’s disease virus.
Several groups failed in their attempts to infect mammalian cell cultures with cell-associated or cell-free MDV and HVT (28,43,59,94,114,128,131). At least 27 cell lines and seven primary cell cultures from 16 species were tested, including Vero and several human cell lines. In many instances, cell lines were examined for the presence of MDV by inoculation of susceptible chickens. In all instances, the chickens remained free of MDV based on the absence of antibodies, viremia, and lesions. However, Bedigian and Sevoian reported the replication of cell-associated and cell-free MDV and HVT in primary hamster kidney (12,13,16) and guinea pig cultures (14,15). Elliott et al. (41) reported the replication of cell-associated HVT in hamster urogenital cell cultures. More recently, Jaikumar et al. (65) published the replication of MDV and HVT in Vero cells but did not indicate if cell-associated or cell-free virus was used to initiate the infection of Vero cells. In no instance were the positive mammalian cells analyzed for MDV by inoculation of susceptible chickens. Witter and Sharma (153) tried to repeat the infection of hamster kidney cells with MDV- or HVT-infected duck embryo fibroblasts (DEF) following the procedures reported by Bedigian and Sevoian (16). It was learned that DEF could survive for at least 12 passages and sustain infection. Elimination of DEF by treatment with anti-DEF serum plus complement abolished the infection. Infection of hamster kidney cells or BHK21 cell line with cell-free HVT could not be repeated (59,94,153). In conclusion, it is the general consensus that mammalian cells are resistant to infection with MDV and HVT (124).
Experimental infection of mammalian species with avian tumor viruses.
Avian retroviruses.
There are a few papers describing the induction of tumors in monkeys with the use of RSV subgroup D viruses. Munroe and Windle (96,97) and others inoculated rhesus monkeys with tumor minces of Carr-Zilber RSV, a subgroup D RSV (96), resulting in sarcomas. However, it could be questioned if these tumors consisted of monkey cells or transplanted chicken sarcoma cells. Subsequently, Deinhardt (38) injected marmoset monkeys with cell-free Carr-Zilber RSV and confirmed that the virus could induce tumors. Karyotyping confirmed that these tumors consisted of transformed marmoset cells. Similar results were reported by two other groups (38,81,84), with the Carr-Zilber strain and Schmidt-Ruppin strain, also a subgroup D RSV (39). Although interesting from an academic point of view, these experiments are not relevant for the discussion on the risks of ALV for human cancer. First of all, these RSV would produce tumors rapidly in chickens and most likely be noticed during the inspection process at the processing plants. Secondly, subgroup D viruses were already extremely rare in the United States prior to the eradication programs for ALV (18,98) and are eradicated from commercial parent flocks as discussed before. We were unable to find any papers on the induction of tumors in mammalian species after infection with ALV-A, ALV-B or REV.
Marek’s disease virus.
A few studies using rodents have been reported. Newborn hamsters inoculated with MDV by intracerebral or subcutaneous injection did not develop tumors or other abnormalities during an 8-mo observation period but were not examined for viremia or antibodies to MDV (36). Over a 6-mo observation period, newborn hamsters and Lewis rats did not develop tumors, viremia, or antibodies to MDV after intraperitoneal injection with cell-associated MDV or MD tumor cells (59). Amos et al. (1) inoculated CFWwd mice intraperitoneally with tumor cells obtained from a field case of MD without checking for the presence of avian retroviruses or confirming that the tumor cells were actually MDVtransformed cells. The diagnosis was made on the presence ofMDVin the FFE, which is inadequate, because most chickens will have MDV in the FFE independently of the presence of tumors. Thirteen of the 67 mice developed tumors that were characterized as mouse cells based on chromosome analysis. MDV could not be rescued by cocultivation of the tumor cells with avian cells. Electron microscopy showed an increase in retrovirus particles that were not further characterized. The authors concluded that ‘‘the etiologic association of MDV and the tumors which arose in the mice is speculative.’’ In our opinion this is a poorly conducted study and does not indicate a proven relationship between MDV and the mouse tumor cells.
Table 1. Summarized data for experimental infections of primates with Marek’s disease virus (MDV) or herpesvirus of turkeys (HVT).
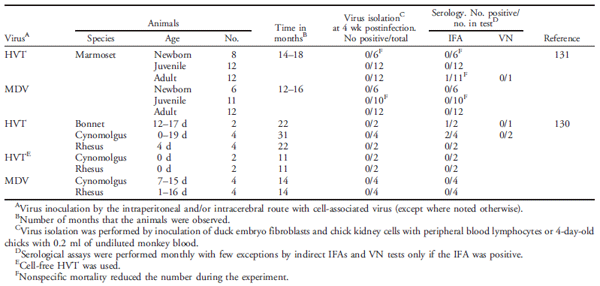
Limited studies have been performed with MDV in primates. Rispens et al. (119) inoculated five adult rhesus monkeys with 100,000 plaque-forming units of CVI-988. In an attempt to cause immunosuppression, the rhesus monkeys were subjected to totalbody X-irradiation of 200 rads 24 hr prior to infection and also received repeated injections with antilymphocyte serum before and after challenge. The monkeys did not develop anti-MDV antibodies or lesions during the 1-yr observation period. Postmortem examination did not reveal gross or microscopic abnormalities. Sharma et al. (130,131) inoculated four species of monkeys with MDV or HVT (see Table 1 for details). None of the monkeys developed viremia or clinical disease. Nonspecific mortality occurred in the marmoset monkeys with a number of spontaneous diseases also observed in noninoculated marmosets. Lesions attributable to HVT or MDV were not observed. In some instances, antibodies to HVT were detected by indirect IFAs in inoculated monkeys as well as in noninfected control monkeys, but VN assays were negative.
Human tumors and avian oncogenic viruses.
There is very little pathological, virological, or molecular evidence of a direct role or even presence of avian tumor viruses in human tumors. Perhaps the biggest experiment with ALV was performed with ALV-contaminated YF vaccines, when large numbers of military troops were vaccinated with YF vaccines. Waters et al. (148) compared 2,659 veterans who died with cancer based on their known YF vaccination status and the year of death, which comprised the periods of 1950– 1954 and 1959–1963. There was no evidence of increased risk of cancer associated with the YF vaccination status of the veterans. Recipients of measles, mumps, and rubella vaccines positive for endogenous ALV and or EAV-0 were negative by RT-PCR for endogenous viral RNA, suggesting an absence of viremia (60,61).
There are no published reports of isolation of MDV from human tumors or other human samples. Recently, real-time quantitative PCR assays as well as nested PCR assays have been used to search for MDV genomic sequences in blood serum or plasma. Laurent et al. (83) examined 202 serum samples by nested PCR for the presence of MDV gD sequences. These samples were previously used for the serological assays reported by Choudat et al. (35). They found 41/ 202 (20.3%) positive samples and sequence analysis indicated that the amplified fragments were identical with the published gD sequence for MDV. There was no significant difference in prevalence between people exposed to poultry versus unexposed people. The same group tested 48 additional sera from people without professional exposure and found 17% positive for MDV gD DNA. These studies were done in a laboratory where no work on MDV had been conducted (Laurent, pers. comm., 2014). They also examined tumor samples from 92 German patients with lymphoma and found 8% positive for gD and 2% positive for MDV meq (Laurent, pers. comm., 2014). These percentages are lower than the percentages positive from people without lymphomas and the importance of these findings is not clear. On the other hand, Hennig et al. (56,57) tested a total of 407 samples and were unable to detect MDV sequences.
To determine if there is any published evidence of involvement of avian retroviruses or MDV in human cancer, we conducted a search with the use of the PubMed data base on March 30, 2014. The search term ‘‘Gene expression profiles in human cancer’’ yielded 10,635 hits, when the search term included ‘‘AND retroviruses’’ the number of hits was reduced to 93. Several of the papers in this list described the activation of human endogenous retrovirus group HERV-K(HML-2) loci (e.g., 47,122,127). Changing the term retrovirus to avian retrovirus, avian leukosis virus, avian sarcoma leukosis virus, or reticuloendotheliosis virus yielded two hits, but these papers discussed the use of avian retrovirus vectors for gene therapy. A similar search using ‘‘Gene expression profiles in human cancer and herpesvirus’’ yielded 100 hits, but these were all related to EBV and KSHV. Changing herpesvirus to Marek’s disease herpesvirus did not show any publications.
Multiple sclerosis and herpesviruses.
The etiology of MS, a debilitating autoimmune demyelinating disorder of the central nervous system in humans, has not been elucidated, but is most likely multifactorial (145). Genetics and different environmental factors are believed to be an important component partly based on the distribution of MS, which is more prevalent in the more northern and southern parts of the northern and southern hemisphere, respectively, than in areas closer to the equator. Virus infections have been linked with MS—especially HHV-6A, although it is not clear if it causes MS or is only associated with MS (106,117,145). MS is characterized by the presence of oligoclonal bands consisting of immunoglobulins in the cerebrospinal fluid. In many instances, these bands react with HHV-6 or EBV (146). The hypothesis that viral infections are involved in the pathogenesis of MS led to the speculation that HVT (21), MDV, or even infectious bursal disease virus could be involved (88,89,90,92,93). The MDV hypothesis was based on a unusual cluster of cases in Key West (FL), which occurred between 1972 and 1986. The researchers linked the presence of large numbers of migratory birds to the MS cases. The migratory birds were supposed to be the source of MDV, but the investigators did not show the presence ofMDV in these birds or explain how these birds would have been infected with MDV. Finally, the researchers did not present any evidence that the MS patients were infected with MDV. To test the MDV hypothesis, Hennig et al. (57) examined peripheral blood lymphocytes (PBL) from 107 MS patients by qPCR for the presence of the MDV UL48 gene. All samples were negative for MDV. As indicated before, those authors also examined 300 samples from healthy volunteer donors for the presence of the MDV gD gene in plasma as well as PBL, which were also negative (56). In conclusion, their results do not support the hypothesis that MDV is involved in the pathogenesis of MS.
CONCLUSIONS
Since the elimination of ALV-A, ALV-B, and ALV-J and REV from poultry breeding stocks to the level of parent flocks and the removal of REV from FPV vaccines,MDV remains as the major avian tumor virus to which people in the industry are exposed. Although it will not be possible to prove that these viruses can never cause cancer in humans, the available literature on ALV, REV, and MDV suggest that these viruses cannot infect humans—let alone be a major factor in the initiation of human cancer. The exception for MDV is the report by Laurent et al. (83), but others were not able to confirm the presence of MDV in human serum or leukocytes (56,57).
Johnson and co-workers have argued for the last 20 yr that avian tumor viruses constitute a significant health risk especially for people working in poultry processing plants (33,34,46,48,67,68,69,70,71). Interestingly to us, workers in the poultry industry (farmers, etc.) were not included in the analysis for the risk of lung cancer (69), while this group certainly would be exposed to all poultry pathogens. The papers we reviewed as examples of this research series indicate a failure to proceed past the hypothesis-generating step, imprecise definitions of risk factors and/or diseases, risk of both information and confounding biases, and large risks of Type-I (false-positive) statistical errors. The actual evidence for the potential to infect humans with exogenous ALV-A and ALV-B or endogenous ALV sequences is lacking—especially in view of the large number of people accidentally inoculated with exogenous and endogenous ALV through contaminated YF and measles vaccines. Moreover, based on the available literature, it needs to be questioned whether humans can be infected with ALV-A or ALV-B. The only ALV subgroups capable of infecting mammalian species (C and D) are at most rarely detected in poultry in the United States and if present in the past, would have been eliminated in parent flocks through the eradication program initiated by the breeding companies in the 1980s.
The presence of antibodies to ALV is likely to represent environmental exposure together with exposure to poultry dust, and does not prove that ALV actually infected humans. Postulating that REV instead of ALV is causing the different array of tumors in poultry requires more evidence than the serological evidence presented by Johnson et al. (73,74). It is not clear why Johnson’s group has not pursued a more rigorous search for the actual presence of ALV, REV, or MDV in humans or collaborated with the numerous groups analyzing gene-expression profiles in human cancer. As mentioned before, this type of gene-expression analysis has not shown any evidence for the presence of avian tumor virus sequences in human tumors.
In conclusion, we believe that the statement by Purchase and Witter (115) remains valid with the inclusion of avian retroviruses: ‘‘Thus in our opinion and in the opinion of experts in the human and avian cancer field, Marek’s disease and vaccines against Marek’s disease pose no human health hazards. Although additional studies are always possible, we believe there is sufficient evidence in this paper to warrant this firm conclusion.’’
REFERENCES
1. Amos, M. A., A. H. Nielsen, and A. A. Werder. Mice inoculated with Marek’s disease tumor cells: increased number of lymphomas. Comp. Immunol. Microbiol. Infect. Dis. 4:21–28. 1981.
2. Anonymous. International Committee on Taxonomy of Viruses. ictvonline.org/virusTaxonomy.asp?taxnode_id520121296. 2014.
3. Astrin, S. M., E. G. Buss, and W. S. Haywards. Endogenous viral genes are non-essential in the chicken. Nature 282:339–341. 1979.
4. Atkins, K. E., A. F. Read, N. J. Savill, K. G. Renz, A. F. Islam, S. W. Walkden-Brown, and M. E. Woolhouse. Vaccination and reduced cohort duration can drive virulence evolution: Marek’s disease virus and industrialized agriculture. Evolution 67:851–860. 2013.
5. Atkins, K. E., A. F. Read, N. J. Savill, K. G. Renz, S. W. Walkden- Brown, and M. E. Woolhouse. Modelling Marek’s disease virus (MDV) infection: parameter estimates for mortality rate and infectiousness. BMC Vet. Res. 7:70. 2011.
6. Bai, J., K. Howes, L. N. Payne, and M. A. Skinner. Sequence of hostrange determinants in the env gene of a full-length, infectious proviral clone of exogenous avian leukosis virus HPRS-103 confirms that it represents a new subgroup (designated J). J. Gen. Virol. 76:181–187. 1995.
7. Bai, J., L. N. Payne, and M. A. Skinner. HPRS-103 (exogenous avian leukosis virus, subgroup J) has an env gene related to those of endogenous elements EAV-0 and E51 and an E element found previously only in sarcoma viruses. J. Virol. 69:779–784. 1995.
8. Baigent, S. J., L. B. Kgosana, A. A. Gamawa, L. P. Smith, A. F. Read, and V. K. Nair. Relationship between levels of very virulent MDV in poultry dust and in feather tips from vaccinated chickens. Avian Dis. 57:440–447. 2013.
9. Barbacid, M., E. Hunter, and S. A. Aaronson. Avian reticuloendotheliosis viruses: evolutionary linkage with mammalian type C retroviruses. J. Virol. 30:508–514. 1979.
10. Barbosa, T., G. Zavala, and S. Cheng. Molecular characterization of three recombinant isolates of avian leukosis virus obtained from contaminated Marek’s disease vaccines. Avian Dis. 52:245–252. 2008.
11. Barnard, R. J. O., D. Elleder, and J. A. T. Young. Avian sarcoma and leukosis virus-receptor interactions: from classical genetics to novel insights into virus–cell membrane fusion. Virology 344:25–29. 2006.
12. Bedigian, H. G., and M. Sevoian. Replication of a herpesvirus from turkeys (HVT) in hamster kidney cell cultures after infection with a cell-free preparation. Avian Dis. 16:1047–1056. 1972.
13. Bedigian, H. G., and M. Sevoian. Susceptibility of mammalian (hamster) cell culture to infection with herpesvirus of turkeys. Appl. Microbiol. 24:275–280. 1972.
14. Bedigian, H. G., and M. Sevoian. Susceptibility of guinea pig cell cultures to infection with cell-bound and cell-free herpesvirus of turkeys. Infect. Immun. 8:482–487. 1973.
15. Bedigian, H. G., and M. Sevoian. Susceptibility of guinea pig cell cultures to infection with cell-bound and cell-free Marek’s disease virus (JM strain). Avian Dis. 17:816–823. 1973.
16. Bedigian, H. G., and M. Sevoian. Susceptibility of mammalian (hamster) cell culture to infection with cellular and cell-free type II leukosis (Marek’s disease) virus. J. Natl. Cancer Inst. 50:129–135. 1973.
17. Birmingham, C. L., D. Dupont, P. Riou, C. Armanet, K. N. Edamura, B. Martinho, A. Serres, S. Jacouton, V. Detrez, B. McNeil, M. Schreiber, D. Gaillac, T. Bonnevay, L. Gisonni-Lex, and L. Mallet. Detection of avian retroviruses in vaccines by amplification on DF-1 cells with immunostaining and fluorescent product-enhanced reverse transcriptase endpoint methods. J. Clin. Microbiol. 51:1496–1504. 2013.
18. Boettiger, D., D. N. Love, and R. A. Weiss. Virus envelope markers in mammalian tropism of avian RNA tumor viruses. J. Virol. 15:108–114. 1975.
19. Bo¨ni, J., J. Stalder, F. Reigel, and J. Schu¨pbach. Detection of reverse transcriptase activity in live attenuated virus vaccines. Clin. Diagn. Virol. 5:43–53. 1996.
20. Borenstein, M., L. V. Hedges, J. P. T. Higgins, and H. R. Rotstein. Introduction to meta-analysis. John Wiley and Sons, Ltd., West Sussex, UK. 2009.
21. Bougiouklis, P. A. Suggesting the possible role of Turkey Herpesvirus or HVT-like as a predisposing factor or causative agent in multiple sclerosis. Med. Hypotheses 67:926–929. 2006.
22. Bova, C. A., J. C. Olsen, and R. Swanstrom. The avian retrovirus env gene family: molecular analysis of host range and antigenic variants. J. Virol. 62:75–83. 1988.
23. Boyce-Jacino, M. T., K. O’Donoghue, and A. J. Faras. Multiple complex families of endogenous retroviruses are highly conserved in the genus Gallus. J. Virol. 66:4919–4129. 1992.
24. Bublot, M., and J. Sharma. Vaccination against Marek’s disease. In: Marek’s disease, an evolving problem. F. Davison, and V. Nair, eds. Elsevier Academic Press, London. pp. 168–185. 2004.
25. Burgess, S. C., and T. F. Davison. Identification of the neoplastically transformed cells in Marek’s disease herpesvirus-induced lymphomas: recognition by the monoclonal antibody AV37. J. Virol. 76:7276–7292. 2002.
26. Calnek, B. W. Marek’s disease: a model for herpesvirus oncology. CRC Crit. Rev. Microbiol. 12:293–320. 1986.
27. Calnek, B. W., H. K. Adldinger, and D. E. Kahn. Feather follicle epithelium: a source of enveloped and infectious cell-free herpesvirus from Marek’s disease. Avian Dis. 14:219–233. 1970.
28. Calnek, B. W., S. H. Madin, and A. J. Kniazeff. Susceptibility of cultured mammalian cells to infection with a herpesvirus from Marek’s disease and T-virus from reticuloendotheliosis of chickens. Am. J. Vet. Res. 30:1403–1412. 1969.
29. Calnek, B. W., K. A. Schat, and J. Fabricant. Modification of Marek’s disease pathogenesis by in ovo infection or prior vaccination. In: Viruses in naturally occurring cancers. M. Essex, G. Todaro, and H. zur Hausen, eds. Cold Spring Harbor, New York. pp. 185–197. 1980.
30. Charman, H. P., R. V. Gilden, and S. Oroszlan. Reticuloendotheliosis virus: detection of immunological relationship to mammalian type C retroviruses. J. Virol. 29:1221–1225. 1979.
31. Chbab, N., A. Egerer, I. Veiga, K. W. Jarosinski, and N. Osterrieder. Viral control of vTR expression is critical for efficient formation and dissemination of lymphoma induced by Marek’s disease virus (MDV). Vet. Res. 41:56. 2010.
32. Cho, B. R. Horizontal transmission of turkey herpesvirus to chickens. IV. Viral maturation in the feather follicle epithelium. Avian Dis. 19:136–141. 1975.
33. Choi, K.-M., and E. S. Johnson. Industrial hygiene assessment of reticuloendotheliosis viruses exposure in the poultry industry. Int. Arch. Occup. Environ. Health 84:375–382. 1975.
34. Choi, K. M., and E. S. Johnson. Occupational exposure assessment using antibody levels: exposure to avian leukosis/sarcoma viruses in the poultry industry. Int. J. Environ. Health Res. 21:306–316. 2011.
35. Choudat, D., G. Dambrine, B. Delemotte, and F. Coudert. Occupational exposure to poultry and prevalence of antibodies against Marek’s disease virus and avian leukosis virus retroviruses. Occup. Environ. Med. 53:403–410. 1996.
36. Churchill, A. E. Herpes-type virus isolated in cell culture from tumors of chickens with Marek’s disease. I. Studies in cell culture. J. Natl. Cancer Inst. 41:939–950. 1968.
37. Davidson, I., and Y. Braverman. Insect contribution to horizontal transmission of reticuloendotheliosis virus. J. Med. Entomol. 42:128–133. 2005.
38. Deinhardt, F. Neoplasms induced by Rous sarcoma virus in new world monkeys. Nature 210:443. 1967.
39. Duff, R. G., and P. K. Vogt. Characteristics of two new avian tumor virus subgroups. Virology 39:18–30. 1969.
40. Dunwiddie, C. T., R. Resnick, M. Boyce-Jacino, J. N. Alegre, and A. J. Faras. Molecular cloning and characterization of gag-, pol-, and envrelated gene sequences in the ev2 chicken. J. Virol. 59:669–675. 1986.
41. Elliott, A. Y., N. Stein, E. E. Fraley, and P. H. Cleveland. Replication of herpesvirus of turkeys in hamster urogenital cell cultures. Am. J. Vet. Res. 34:427–429. 1973.
42. Enrietto, P. J., and M. J. Hayman. Structure and virus-associated oncogenes of avian sarcoma and leukemia viruses. In: Avian Leukosis. G. F. de Boer, ed. Martinus Nijhoff, Boston, MA. pp. 29–46. 1987.
43. Esposito, J., P. D. Lukert, and C. S. Eidson. Marek’s disease herpesvirus: growth and detection in vivo and in vitro. Am. J. Vet. Res. 33:171–175. 1972.
44. Fadly, A., R. Silva, H. Hunt, A. Pandiri, and C. Davis. Isolation and characterization of an adventitious avian leukosis virus isolated from commercial Marek’s disease vaccines. Avian Dis. 50:380–385. 2006.
45. Fakhrul Islam, A. F., S. W. Walkden-Brown, P. J. Groves, and G. J. Underwood. Kinetics of Marek’s disease virus (MDV) infection in broiler chickens 1: effect of varying vaccination to challenge interval on vaccinal protection and load of MDV and herpesvirus of turkey in the spleen and feather dander over time. Avian Pathol. 37:225–235. 2008.
46. Felini, M., E. A. Johnson, N. Preacely, V. Sarda, H. Ndetan, and S. Bangara. A pilot case-cohort study of liver and pancreatic cancers in poultry workers. Ann. Epidemiol. 21:755–766. 2011.
47. Flockerzi, A., A. Ruggieri, O. Frank, M. Sauter, E. Maldener, B. Kopper, B. Wullich, W. Seifarth, N. Muller-Lantzsch, C. Leib-Mosch, E. Meese, and J. Mayer. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics 9:354. 2008.
48. Gandhi, S., M. J. Felini, H. Ndetan, K. Cardarelli, S. Jadhav, M. Faramawi, and E. S. Johnson. A pilot case-cohort study of brain cancer in poultry and control workers. Nutr. Cancer 66:343–350. 2014.
49. Garcia, M., N. Narang, W. M. Reed, and A. M. Fadly. Molecular characterization of reticuloendotheliosis virus insertions in the genome of field and vaccine strains of fowl poxvirus. Avian Dis. 47:343–354. 2003.
50. Gautier, R., A. Jiang, V. Rousseau, R. Dornburg, and T. Jaffredo. Avian reticuloendotheliosis virus strain A and spleen necrosis virus do not infect human cells. J. Virol. 74:518–522. 2000.
51. Gavora, J. S. Influences of avian leukosis virus infection on production and mortality and the role of genetic selection in the control of lymphoid leukosis. In: Avian leukosis. G. F. de Boer, ed. Martinus Nijhoff, Boston, MA. pp. 241–260. 1987.
52. Gimeno, I. M. Marek’s disease vaccines: a solution for today but a worry for tomorrow? Vaccine 26:C31–C41. 2008.
53. Gingerich, E., R. E. Porter, B. Lupiani, and A. M. Fadly. Diagnosis of myeloid leukosis induced by a recombinant avian leukosis virus in commercial white leghorn egg laying flocks. Avian Dis. 46:745–748. 2002.
54. Grachev, V., D. Magrath, and E. Griffiths. Requirements for yellow fever vaccine. In: WHO Technical Report No. 872. WHO, Geneva, Switzerland. pp. 31–47. 1998.
55. Harris, R. J., R. M. Dougherty, P. M. Biggs, L. N. Payne, A. P. Goffe, A. E. Churchill, and R. Mortimer. Contaminant viruses in two live virus vaccines produced in chick cells. J. Hyg. (Lond.) 64:1–7. 1966.
56. Hennig, H., N. Osterrieder, M. Muller-Steinhardt, H. M. Teichert, H. Kirchner, and K. P. Wandinger. Detection of Marek’s disease virus DNA in chicken but not in human plasma. J. Clin. Microbiol. 41:2428–2432. 2003.
57. Hennig, H., K. Wessel, P. Sondermeijer, H. Kirchner, and K. P. Wandinger. Lack of evidence for Marek’s disease virus genomic sequences in leukocyte DNA from multiple sclerosis patients in Germany. Neurosci. Lett. 250:138–140. 1998.
58. Hertig, C., B. E. Coupar, A. R. Gould, and D. B. Boyle. Field and vaccine strains of fowlpox virus carry integrated sequences from the avian retrovirus, reticuloendotheliosis virus. Virology 235:367–376. 1997.
59. Hlozanek, I., and V. Sovova. Lack of pathogenicity of Marek’s disease herpesvirus and herpesvirus of turkeys for mammalian hosts and mammalian cell cultures. Folia Biol. 20:51–58. 1974.
60. Hussain, A. I., J. A. Johnson, M. Da Silva Freire, and W. Heneine. Identification and characterization of avian retroviruses in chicken embryoderived yellow fever vaccines: investigation of transmission to vaccine recipients. J. Virol. 77:1105–1111. 2003.
61. Hussain, A. I., V. Shanmugam, W. M. Switzer, S. X. Tsang, A. Fadly, D. Thea, R. Helfand, W. J. Bellini, T. M. Folks, and W. Heneine. Lack of evidence of endogenous avian leukosis virus and endogenous avian retrovirus transmission to measles, mumps, and rubella vaccine recipients. Emerg. Infect. Dis. 7:66–72. 2001.
62. Ioannidis, J. P. A. Why most published research findings are false. PLoS Med. 2:0696–0700. 2005.
63. Isfort, R., D. Jones, R. Kost, R. Witter, and H.-J. Kung. Retrovirus insertion into herpesvirus in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 89:991–995. 1992.
64. Jacobson, R. H. Validation of serological assays for diagnosis of infectious diseases. Rev. Sci. Tech. Off. Int. Epizoot. 17:469–526. 1998.
65. Jaikumar, D., K. M. Read, and G. A. Tannock. Adaptation of Marek’s disease virus to the Vero continuous cell line. Vet. Microbiol. 79:75–82. 2001.
66. Jarosinski, K. W., and K. A. Schat. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: the importance of vIL-8 exon I. Virus Genes 34:9–22. 2007.
67. Johnson, E. S. Poultry oncogenic retroviruses and humans. Cancer Detect. Prev. 18:9–30. 1994.
68. Johnson, E. S. Cancer mortality in workers employed in cattle, pigs, and sheep slaughtering and processing plants. Environ. Int. 37:950–959. 2011.
69. Johnson, E. S., and K.-M. Choi. Lung cancer risk in workers in the meat and poultry industries—a review. Zoonoses Public Health 59:303–313. 2012.
70. Johnson, E. S., and C. M. Griswold. Oncogenic retroviruses of cattle, chickens and turkeys: potential infectivity and oncogenicity for humans. Med. Hypotheses 46:354–356. 1996.
71. Johnson, E. S., and H. Ndetan. Non-cancer mortality in poultry slaughtering/processing plant workers belonging to a union pension fund. Environ. Int. 37:322–327. 2011.
72. Johnson, E. S., H. Ndetan, and K.-M. Lo. Cancer mortality in poultry slaughtering/processing plant workers belonging to a union pension fund. Environ. Res. 110:588–594. 2010.
73. Johnson, E. S., L. G. Nicholson, and D. T. Durack. Detection of antibodies to avian leukosis/sarcoma viruses (ALSV) and reticuloendotheliosis viruses (REV) in humans by ELISA. Cancer Detect. Prev. 19:394–404. 1995.
74. Johnson, E. S., L. Overby, and R. Philpot. Detection of antibodies to avian leukosis/sarcoma viruses and reticuloendotheliosis viruses in humans by western blot assay. Cancer Detect. Prev. 19:472–486. 1995.
75. Kalland, K. H., X. S. Ke, and A. M. Oyan. Tumour virology— history, status and future challenges. APMIS 117:382–399. 2009.
76. Kato, S., K. Ono, M. Naito, and S. Tanabe. Immunological studies on Marek’s disease virus and Epstein-Barr virus. In: Oncogenesis and herpesvirues. P. M. Biggs, G. de-The, and L. N. Payne, eds. International Agency for Research on Cancer, Lyon, France. pp. 485–488. 1972.
77. Keshet, E., and H. M. Temin. Cell killing by spleen necrosis virus is correlated with a transient accumulation of spleen necrosis virus DNA. J. Virol. 31:376–388. 1979.
78. Kingham, B. F., V. Zelnik, J. Kopacek, V. Majerciak, E. Ney, and C. J. Schmidt. The genome of herpesvirus of turkeys: comparative analysis with Marek’s disease viruses. J. Gen. Virol. 82:1123–1135. 2001.
79. Koo, H. M., A. M. Brown, Y. Ron, and J. P. Dougherty. Spleen necrosis virus, an avian retrovirus, can infect primate cells. J. Virol. 65:4769–4776. 1991.
80. Kost, R., D. Jones, R. Isfort, R. Witter, and H. J. Kung. Retrovirus insertion into herpesvirus: characterization of a Marek’s disease virus harboring a solo LTR. Virology 192:161–169. 1993.
81. Kumanishi, T., F. Ikuta, K. Nishida, K. Ueki, and T. Yamamoto. Brain tumors induced in adult monkeys by Schmidt-Ruppin strain of Rous sarcoma virus. Gann 64:641–643. 1973.
82. Kung, H.-J., and N. J. Maihle. Molecular basis of oncogenesis by non-acute avian retroviruses. In: Avian leukosis. G. F. de Boer, ed. Martinus Nijhoff, Boston, MA. pp. 77–100. 1987.
83. Laurent, S., E. Esnault, G. Dambrine, A. Goudeau, D. Choudat, and D. Rasschaert. Detection of avian oncogenic Marek’s disease herpesvirus DNA in human sera. J. Gen. Virol. 82:233–240. 2001.
84. Levy, B. M., A. C. Taylor, S. Hampton, and G. W. Thoma. Tumors of the marmoset produced by Rous sarcoma virus. Cancer Res. 29:2237–2248. 1969.
85. Li, L., and F. J. Hoerr. Diagnosis of ALV-J by PCR. Archives of Thompson-Bishop-Sparks State Diagnostic Laboratory, Auburn, AL. 2014.
86. Liu, J. L., L. F. Lee, Y. Ye, Z. Qian, and H. J. Kung. Nucleolar and nuclear localization properties of a herpesvirus bZIP oncoprotein, MEQ. J. Virol. 71:3188–3196. 1997.
87. Liu, Q., J. Zhao, J. Su, J. Pu, G. Zhang, and J. Liu. Full genome sequences of two reticuloendotheliosis viruses contaminating commercial vaccines. Avian Dis. 53:341–346. 2009.
88. MacGregor, H. S., and Q. I. Latiwonk. Search for the origin of multiple sclerosis by first identifing the vector. Med. Hypotheses 37:67–73. 1992.
89. MacGregor, H. S., and Q. I. Latiwonk. Complex role of gammaherpesviruses in multiple sclerosis and infectious mononucleosis. Neurol. Res. 15:391–394. 1993.
90. MacGregor, H. S., and Q. I. Latiwonk. Is MS an auto-immune disease or a chronic gammaherpesvirus infection? J. Clin. Lab. Immunol. 48:45–74. 1996.
91. Markham, F. S., and S. Levine. The absence of serologic responses by children and adults to avian leukosis virus in measles vaccine. Arch. Gesamte Virusforsch. 16:305–310. 1965.
92. McHatters, G. R., and R. G. Scham. Bird viruses in multiple sclerosis: combination of viruses or Marek’s alone? Neurosci. Lett. 188:75–76. 1995.
93. McStreet, G. H., R. B. Elkunk, and Q. I. Latiwonk. Investigations of environmental conditions during cluster indicate probable vectors of unknown exogenous agent(s) of multiple sclerosis. Comp. Immunol. Microbiol. Infect. Dis. 15:75–77. 1992.
94. Meulemans, G., P. Halen, and P. Schyns. Susceptibility of mammalian and avian cell cultures to infection with cell-free turkey herpes virus. J. Comp. Pathol. 83:605–608. 1973.
95. Moher, D. A., A. Liberati, J. Tetzlaff, D. G. Altman, and P. Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann. Intern. Med. 151:1–7. 2009.
96. Munroe, J. S., and W. F. Windle. Tumors induced in primates by chicken sarcoma virus. Science 140:1415–1416. 1964.
97. Munroe, J. S., and W. F. Windle. Tumours induced by chicken sarcoma in monkeys conditioned with progesterone. Nature 216:811–813. 1967.
98. Nair, V., and A. M. Fadly. Leukosis/sarcoma group. In: Diseases of poultry, 13th ed. D. E. Swayne, J. R. Glisson, L. R. McDougald, J. V. Nolan, D. L. Suarez, and V. Nair, eds. Wiley-Blackwell, Ames, IA. pp. 553–592. 2013.
99. Nair, V., and H. J. Kung. Marek’s disease virus oncogenicity: molecular mechanisms. In: Marek’s disease, an evolving problem. F. Davison, and V. Nair, eds. Elsevier Academic Press, Oxford, England. pp. 32–48. 2004.
100. Nair, V., G. Zavala, and A. M. Fadly. Reticuloendotheliosis. In: Diseases of poultry, 13th ed. D. E. Swayne, J. R. Glisson, L. R. McDougald, J. V. Nolan, D. L. Suarez, and V. Nair, eds. Wiley-Blackwell, Ames, IA. pp. 593–604. 2013.
101. Naito, M., K. Ono, T. Doi, and S. Kato. Antibodies in human and monkey sera to herpes-type virus from a chicken with Marek’s disease and to EB virus detected by the immunofluorescence test. Biken J. 14:161–166. 1971.
102. Naito, M., K. Ono, S. Tanabe, T. Doi, and S. Kato. Detection in chicken and human sera of antibody against herpes type virus from a chicken with Marek’s disease virus and EB virus demonstrated by the indirect immunofluorescence test. Biken J. 13:205–212. 1970.
103. Nakajima, K., C. H. Kweon, S. H. An, T. Shibayama, N. Wakamiya, S. Kato, and K. Hirai. Identification of the cross-reactive antigens between Marek’s disease virus and pseudorabies virus by monoclonal antibodies. Avian Dis. 34:479–484. 1990.
104. Niewiadomska, A. M., and R. J. Gifford. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 11:e1001642. 2013.
105. Nuzzo, R. Statistical errors. Nature 506:150–152. 2014.
106. Olival, G. S., B. M. Lima, L. M. Sumita, V. Serafim, M. C. Fink, L. H. Nali, C. M. Romano, R. B. Thomaz, V. B. Cavenaghi, C. P. Tilbery, and A. C. Penalva-de-Oliveira. Multiple sclerosis and herpesvirus interaction. Arq. Neuropsiquiatr. 71:727–730. 2013.
107. Ono, K., S. Tanabe, M. Naito, T. Doi, and S. Kato. Antigen common to a herpes type virus from chickens with Marek’s disease and EB virus from Burkitt’s lymphoma cells. Biken J. 13:213–217. 1970.
108. Oroszlan, S., M. Barbacid, T. D. Copeland, S. A. Aaronson, and R. V. Gilden. Chemical and immunological characterization of the major structural protein (p28) of MMC-1, a rhesus monkey endogenous type C virus: homology with the major structural protein of avian reticuloendotheliosis virus. J. Virol. 39:845–854. 1981.
109. Parcells, M. S., S. F. Lin, R. L. Dienglewicz, V. Majerciak, D. R. Robinson, H. C. Chen, Z. Wu, G. R. Dubyak, P. Brunovskis, H. D. Hunt, L. F. Lee, and H. J. Kung. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 75:5159–5173. 2001.
110. Payne, L. N., S. R. Brown, N. Bumstead, K. Howes, J. A. Frazier, and M. E. Thouless. A novel subgroup of exogenous avian leukosis virus in chickens. J. Gen. Virol. 72:801–807. 1991.
111. Payne, L. N., and V. Nair. The long view: 40 years of avian leukosis research. Avian Pathol. 41:11–19. 2012.
112. Pham, T. D., J. L. Spencer, and E. S. Johnson. Detection of avian leukosis virus in albumen of chicken eggs using reverse transcription polymerase chain reaction. J. Virol. Methods 78:1–11. 1999.
113. Piraino, F., E. R. Krumbiegel, and H. J. Wisniewski. Serologic survey of man for avian leukosis virus infection. J. Immunol. 98:702–706. 1967.
114. Purchase, H. G., B. R. Burmester, and C. H. Cunningham. Responses of cell cultures from various avian species to Marek’s disease virus and herpesvirus of turkeys. Am. J. Vet. Res. 32:1811–1823. 1971.
115. Purchase, H. G., and R. L. Witter. Public health concerns from human exposure to oncogenic avian herpesviruses. J. Am. Vet. Med. Assoc. 189:1430–1436. 1986.
116. Qian, Z., P. Brunovskis, F. Rauscher 3rd, L. Lee, and H. J. Kung. Transactivation activity of Meq, a Marek’s disease herpesvirus bZIP protein persistently expressed in latently infected transformed T cells. J. Virol. 69:4037–4044. 1995.
117. Reynaud, J. M., and B. Horvat. Animal models for human herpesvirus 6 infection. Front. Microbiol. 4:174. 2013.
118. Richman, A. V., C. G. Aulisio, W. G. Jahnes, and N. M. Tauraso. Avian leukosis antibody response in individuals given chicken embryo derived vaccines. Proc. Soc. Exp. Biol. Med. 139:235–237. 1972.
119. Rispens, B. H., H. van Vloten, N. Mastenbroek, H. J. Maas, and K. A. Schat. Control of Marek’s disease in the Netherlands. I. Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis. 16:108–125. 1972.
120. Ross, N., G. O’Sullivan, C. Rothwell, G. Smith, S. C. Burgess, M. Rennie, L. F. Lee, and T. F. Davison. Marek’s disease virus EcoRI-Q gene (meq) and a small RNA antisense to ICP4 are abundantly expressed in CD4+ cells and cells carrying a novel lymphoid marker, AV37, in Marek’s disease lymphomas. J. Gen. Virol. 78:2191–2198. 1997.
121. Roth, F. K., and R. M. Dougherty. Search for group-specific antibodies of avian leukosis virus in human leukemic sera. J. Natl. Cancer. Inst. 46:1357–1359. 1971.
122. Ruprecht, K., H. Ferreira, A. Flockerzi, S. Wahl, M. Sauter, J. Mayer, and N. Mueller-Lantzsch. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J. Virol. 82:10008– 10016. 2008.
123. Schat, K. A. Role of the spleen in the pathogenesis of Marek’s disease. Avian Pathol. 10:171–182. 1980.
124. Schat, K. A. Characteristics of the virus. In: Marek’s disease. L. N. Payne, ed. Martinus Nijhoff, Boston. pp. 77–112. 1985.
125. Schat, K. A., and B. W. Calnek. Characterization of an apparently nononcogenic Marek’s disease virus. J. Natl. Cancer Inst. 60:1075–1082. 1978.
126. Schat, K. A., and V. Nair. Marek’s disease. In: Diseases of poultry, 13th ed. D. E. Swayne, J. R. Glisson, L. R. McDougald, J. V. Nolan, D. L. Suarez, and V. Nair, eds. Wiley-Blackwell, Ames, IA. pp. 515–552. 2013.
127. Schmitt, K., J. Reichrath, A. Roesch, E. Meese, and J. Mayer. Transcriptional profiling of human endogenous retrovirus group HERVK( HML-2) loci in melanoma. Genome Biol. Evol. 5:307–328. 2013.
128. Sharma, J. M. In vitro cell association of Marek’s disease herpesvirus. Am. J. Vet. Res. 32:291–301. 1971.
129. Sharma, J. M., D. Burger, and S. G. Kenzy. Serological relationships among herpesviruses: Cross-reaction between Marek’s disease virus and pseudorabies virus as detected by immunofluorescence. Infect. Immun. 5:406–411. 1972.
130. Sharma, J. M., R. L. Witter, B. R. Burmester, and J. C. Landon. Public health implications of Marek’s disease virus and herpesvirus of turkeys. Studies on human and subhuman primates. J. Natl. Cancer Inst. 51:1123–1128. 1973.
131. Sharma, J. M., R. L. Witter, G. Shramek, L. G. Wolfe, B. R. Burmester, and F. Deinhardt. Lack of pathogenicity of Marek’s disease virus and herpesvirus of turkeys in marmoset monkeys. J. Natl. Cancer Inst. 49:1191–1197. 1972.
132. Shott, S. Testing ideas and estimating clinical importance. J. Am. Vet. Med. Assoc. 238:871–876. 2011.
133. Smith, E. J. Endogenous avian leukemia viruses. In: Avian leukosis. G. F. de Boer, ed. Martinus Nijhoff, Boston, MA. pp. 101–120. 1987.
134. Smith, L. M., S. R. Brown, K. Howes, S. McLeod, S. S. Arshad, G. S. Barron, K. Venugopal, J. C. McKay, and L. N. Payne. Development and application of polymerase chain reaction (PCR) tests for the detection of subgroup J avian leukosis virus. Virus Res. 54:87–98. 1998.
135. Spatz, S. J., L. Petherbridge, Y. Zhao, and V. Nair. Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J. Gen. Virol. 88:1080–1096. 2007.
136. Spatz, S. J., and K. A. Schat. Comparative genomic sequence analysis of the Marek’s disease vaccine strain SB-1. Virus Genes 42:331–338. 2011.
137. Spencer, J. L. Progress towards eradication of lymphoid leukosis viruses—a review. Avian Pathol. 13:599–619. 1984.
138. Spencer, J. L., L. B. Crittenden, B. R. Burmester, C. Romero, and R. L. Witter. Lymphoid leukosis viruses and GS antigen in unincubated chicken eggs. Avian Pathol. 5:221–226. 1976.
139. Stroup, D. F., J. A. Berlin, S. C. Morton, I. Olkin, G. D. Williamson, D. Rennie, D. Moher, B. J. Becker, T. A. Sipe, and S. B. Tucker. Meta-analysis of observational studies in epidemiology: a proposal for reporting. J. Am. Med. Assoc. 283:2008–2012. 2000.
140. Tischer, B. K., and N. Osterrieder. Herpesviruses—a zoonotic threat? Vet. Microbiol. 140:266–270. 2010.
141. Trapp, S., M. S. Parcells, J. P. Kamil, D. Schumacher, B. K. Tischer, P. M. Kumar, V. K. Nair, and N. Osterrieder. A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J. Exp. Med. 203:1307–1317. 2006.
142. Tsang, S. X., W. M. Switzer, V. Shanmugam, J. A. Johnson, C. Goldsmith, A. Wright, A. Fadly, D. Thea, H. Jaffe, T. M. Folks, and W. Heneine. Evidence of avian leukosis virus subgroup E and endogenous avian virus in measles and mumps vaccines derived from chicken cells: investigation of transmission to vaccine recipients. J. Virol. 73:5843–5851. 1999.
143. U.S. Department of Agriculture, SAM-415. Supplemental assay methods for detecting lymphoid leukosis biocontamination by the COFAL test. Biologics Laboratories, Animal Plant and Health Inspection Service, Ames, IA. 2005.
144. U.S. Department of Agriculture. Veterinary Service Memorandum No. 800.88. Testing for reticuloendotheliosis virus contamination. Center for Veterinary Biologic, and Licensing, Ames, IA. pp. 1–4. 1999.
145. Venkatesan, A., and R. T. Johnson. Infections and multiple sclerosis. In: Handbook of Clinical Neurology 2014/02/11. D. S. Goodin, ed. Elsevier, London. pp. 151–171. 2014.
146. Virtanen, J. O., J. Wohler, K. Fenton, D. S. Reich, and S. Jacobson. Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult. Scler. J. 20:27–34. 2014.
147. Watanabe, S., and H. M. Temin. Construction of a helper cell line for avian reticuloendotheliosis virus cloning vectors. Mol. Cell. Biol. 3:2241–2249. 1983.
148. Waters, T. D., P. S. Anderson Jr., G. W. Beebe, and R. W. Miller. Yellow fever vaccination, avian leukosis virus, and cancer risk in man. Science 177:76–77. 1972.
149. Weissmahr, R. N., J. Schupbach, and J. Boni. Reverse transcriptase activity in chicken embryo fibroblast culture supernatants is associated with particles containing endogenous avian retrovirus EAV-0 RNA. J. Virol. 71:3005–3012. 1997.
150. Williams, S. M., T. Barbosa, S. Hafner, and G. Zavala. Myxosarcomas associated with avian leukosis virus subgroup A infection in fancy breed chickens. Avian Dis. 54:1319–1322. 2010.
151. Witter, R. L. Turkey herpesvirus: lack of oncogenicity for turkeys. Avian Dis. 16:666–670. 1972.
152. Witter, R. L. Increased virulence of Marek’s disease virus field isolates. Avian Dis. 41:149–163. 1997.
153. Witter, R. L., and J. M. Sharma. Transient infectivity and heterokaryon formation in hamster cell cultures inoculated with cellassociated stocks of Marek’s disease virus and herpesvirus of turkeys. J. Natl. Cancer Inst. 53:1731–1742. 1974.
154. Witter, R. L., and J. J. Solomon. Experimental infection of turkeys and chickens with a herpesvirus of turkeys (HVT). Avian Dis. 16:34–44. 1972.
155. Zavala, G., and S. Cheng. Detection and characterization of avian leukosis virus in Marek’s disease vaccines. Avian Dis. 50:209–215. 2006.
156. Zavala, G., B. Lucio-Martinez, S. Cheng, and T. Barbosa. Sarcomas and myelocytomas induced by a retrovirus related to myeloblastosisassociated virus type 1 in white leghorn egg layer chickens. Avian Dis. 50:201–208. 2006.
ACKNOWLEDGMENTS
The authors want to thank Beth Licitra for performing the BLAST searches and are grateful for the critical review of the manuscript by Dr. Guillermo Zavala.
Financial disclosure. The authors were asked by the United States Poultry and Egg Association (USPOULTRY) to write this review and were financially compensated for their effort. USPOULTRY did not review, edit, or influence the contents of this article. The article solely reflects the opinion of the authors.
Related topics:
Authors:

Cornell University
Recommend
Comment
Share

25 de mayo de 2015
Well the issue of transmission of disease from avian to human being is a new culture spread by some world wide due to various thinking at different levels including some scientists. The fear generating concepts in business management has become the present age trend. Well even there is small issues like water purifiers are also marketed with advantage arguments between firms and all most all marketing items are based on emotional and fear concepts or linked with profit generation. If i say AI is also can be grouped in this category of concept then perhaps many scientists do not agree with reasons. But i agree with the concept that non of the Avian disease can make fatal issues to human and human to human transmission of poultry disease is thought without findings. Present paper do say it is a review but it is a fact. AI has become the issue of the world now and lot of work and expenditures are taken care by all continents with concept to control or save human race. vaccines, bio security items,costly antibiotics, AI administration etc are ok but I have worked in poultry for the last 40 years and i do not know how to fear poultry and why?All animals and birds have independent systems due to super natural powers beyond our understanding but still there is no harm to take care all points raised at different levels for poultry food productions by experienced producers. This is my personal opinion on viral diseases of poultry. I enjoyed the article which is placed here. Nice review on poultry oncogenic virus. Dr m rath
Recommend
Reply


25 de mayo de 2015
Thanks Sir for this article
Sir, I would ask help in suggesting idea of proposal project with details to study for a doctorate in poultry diseases If you are interested to answer , email me please : montaqm12@yahoo.com
Thank you again
Recommend
Reply

Would you like to discuss another topic? Create a new post to engage with experts in the community.







You may be interested in
