INTRODUCTION
The poultry gastrointestinal tract (GIT) is home to a complex, dynamic, and variable bacterial-dominated microbiota (Zhu et al., 2002). This GIT microbiome variation may be explained by different host characteristics and environmental factors (Kers et al., 2018), including birds age (Ballou et al., 2016; Pedroso et al., 2016), sex (Torok et al., 2013; Zhao et al., 2013), type and breed (Videnska et al., 2014; Kim et al., 2015), and GIT regions (Yeoman et al., 2012). Of the external factors that influence the microbiota composition, the diet composition (Pan and Yu, 2014; Walugembe et al., 2015) and the use of feed additives (Videnska et al., 2013; Costa et al., 2017) have been well-studied, but focus has been on the effects of different protein/fiber sources, growth promotion antibiotics, or pre/probiotic antibiotic alternatives compared to standard corn/soy-based diets.
Soybean and its byproducts (especially soybean meal) are ingredients normally found in animal rations due to their elevated levels of protein and several essential amino acids (National Research Council–NRC, 1994). Soybean is typically a major component of chicken rations in both conventional and alternative poultry production systems (Husak et al., 2008), and certain compounds found in soybeans such as isoflavones have been regarded as beneficial to human health (Setchell, 1998; Setchell and Cassidy, 1999). The popular claim that “you are what you eat” is supported by science. As an illustration, isoflavones have been detected in both the tissues and egg yolk of birds consuming soybean meal (Galdos et al., 2009).
Despite the health benefits associated with the intake of soybeans, in recent years the effects of soy-based foods on human health has become increasingly controversial among the general public (D’Adamo and Sahin, 2014). Moreover, soybean allergy has become a relatively common condition in humans, especially in children (Savage et al., 2010), and an increasing number of consumers are seeking soy-free products. Some consumers now demand that products such as eggs and meat must come from chickens fed soy-free diets. In order to accommodate that demand, several companies now offer soy-free feeds, which are marketed to both the conventional and alternative (e.g., organic, pasture-raised) chicken production systems.
Since feeding soy-free diets has become a common practice among pasture-raised chicken producers (with many conventional operations following suit), we aimed to study the effects of feeding 2 different diets—one with soybean and another free of soy—on the intestinal microbiome of pastureraised chickens. Using a farm-to-fork approach, samples were collected at several stages of the production chain starting with the live birds (fecal samples), during the carcass processing stages [when cecal contents and whole carcass rinses (WCR) were obtained], and also after keeping the carcasses frozen for 1 month (when a final product WCR was obtained). All samples collected were subjected to 16s rRNA gene sequencing for assessment of their microbial populations. We hypothesized that birds in the 2 different feed regimens would develop substantially different microbiomes. Furthermore, we wanted to investigate if such microbiomes had significant differences regarding important foodborne pathogen groups such as Salmonella, Campylobacter, and Acinetobacter, which would indicate that their numbers can be modulated by birds’ diet.
MATERIALS AND METHODS
This study was exempt from animal ethics approval since a commercial farm managed and butchered all birds, thus, ethics approval was not required as per applicable institutional and national guidelines and regulations. Briefly, the cecal samples and WCR were collected after birds had already been processed. The only samples that were collected while birds were still alive were the fecal material from their pastures, which were obtained immediately after they left the paddocks.
Animals, Diets, and Sampling Methods
This study was conducted in one pasture-raised broiler farm located in the southeastern region of the United States. Over a period of 2 years, 5 flocks of pasture-raised chickens were raised in this system−3 of those received the soybean-based (SB) feed, while the other 2 flocks were fed with the soy-free feed (SF). Except for the type of supplement offered, all the 5 flocks were raised on the same farm and under the same conditions. For all flocks, 1-day-old Freedom Ranger type chick were purchased from the same hatchery flock (Freedom Ranger Hatchery, Reinholds, PA, USA), and during the brooding period (0–∼4 weeks of age), chicks were reared under heat lamps with bedding material composed of wood shavings (with fresh shaving added weekly on top of the existing bedding). At ∼4 weeks of age, young broilers were transferred to pasture under modified Salatin-type chicken tractor housing systems (3 houses per flock with ∼125 birds per house). During live production, broilers were provided feed and water ad libitum, and the tractors were moved onto a fresh piece of pasture every day. As broilers increased in size, plastic temporary fencing was placed around the tractors to allow for increased grazing area, and this fencing was moved daily with the houses. At ∼12 weeks of age, broilers were transported to a USDA-inspected facility where processing samples were collected. Additionally, processed carcasses were collected from the facility and stored frozen for 1 month to represent the final product this farmer typically provides to consumers. After this period of storage, WCR of the final product were obtained.
Fecal samples were collected from the pasture where the flock was residing at the time of sampling. Samplings occurred three times during grow-out: (i) within a few days of being placed in the pasture (∼4 weeks of age), (ii) halfway through their time on pasture (∼7 weeks of age), and (iii) on the day the flock was processed (∼12 weeks of age). At each sampling time, the pasture area was divided into five separate sections, and five subsamples in each section were pooled into a single sample for each section (a total of five fecal samples were collected on each sampling day). Samples were collected from fresh droppings on the soil surface. Gloves were changed between each sampling area.
Once birds arrived the USDA-inspected slaughter house, the processing samples were collected. They consisted of cecal contents and WCR. Upon evisceration, cecal sacs from five carcasses were removed and placed into a single sampling bag to create a pooled sample. A total of five pooled samples (n = 25) were created. Gloves and scissors were changed between each pooled sample. Prior to packaging and storage of the carcasses for the consumer, each of the 25 carcasses were placed in sterile plastic bags, rinsed with 100 ml of 10 mM phosphate-buffered saline (PBS) and vigorously shaken for 1 min. Whole carcass rinses from five carcasses were pooled together in a filtered stomacher bag creating five pooled samples (n = 25). Carcasses were returned to the processor to be packed and stored according to the usual in-plant procedure. Based on a farm management survey, the average storage temperate (−20°C) and time (∼4 weeks) were emulated for the final product samples, and the procedure described above was repeated 1 month later to obtain the final product WCR samples.
All fecal, cecal, and WCR samples were transported back to the laboratory on ice and processed within 2 h of collection. To prepare the environmental samples for homogenization, 3 g of feces or 5 ceca were combined within filtered stomacher bags (Seward Laboratory Systems, Inc., Davie, FL), and diluted 1:3 using 10 mM PBS. For the WCR, 100 ml of 10 mM PBS were added to each carcass within the storage bag, and the bags were vigorously shaken for 60 s. Five WCR were pooled into a single filtered stomacher bag, and this was repeated a total of 5 times (n = 25 carcass rinses). No further dilution in 10 mM PBS was required for the WCR samples. All samples were homogenized for 60 s and these homogenates were used for DNA extraction.
DNA Extractions
DNA extractions were performed using 0.33 g of feces, 0.5 ml of cecal homogenate, and 0.5 ml of WCR. DNA was extracted from samples according to a semi-automated hybrid DNA extraction protocol previously described (Rothrock et al., 2014). This method was a combination of a mechanical method using the FastDNA Spin Kit for Feces (MP Biomedicals, Solon, OH, USA) and an enzymatic method based on the QIAamp DNA Stool Mini Kit (QIAGEN, Valencia, CA). DNA purification was performed using the DNA Stool—Human Stool—Pathogen Detection Protocol of the QIAcube Robotic Workstation. After purification, the DNA concentration in each sample was determined spectrophotometrically using the Take3 plate in conjunction with the Synergy H4 multimode plate reader (BioTek, Winooski, VT).
16S rRNA Gene Sequencing and Analysis
Library construction and sequencing were performed by the Earth Microbiome Project Laboratory at the U.S. Department of Energy, Argonne National Laboratory (Argonne, IL) according to their standard protocols (http://www.earthmicrobiome. org/protocols-and-standards/16s/). The number of PCR cycles for library construction was 35. The hypervariable V4 domain of bacterial 16S rRNA gene was amplified using the F515 (5′ -CACGGTCGKCGGCGCCATT-3′ ) and R806 (5′ - GGACTACHVGGGTWTCT AAT-3′ ) primer set with each primer containing Illumina adapter regions (Illumina, Inc., San Diego, CA) and the reverse primer containing the Golay barcodes to facilitate multiplexing (Caporaso et al., 2011). The initial demultiplexing and quality filtering steps were performed using the QIIME v1.9.1 (Quantitative Insights Into Microbial Ecology) pipeline (Caporaso et al., 2010b). The 16S rRNA gene amplicon pool was demultiplexed by internal barcodes to identify individual samples and primers were removed using the split_library_fastq.py script. Sequences were chimera checked against the Greengenes 13_8 database (DeSantis et al., 2006) and clustered into Operational Taxonomic Units (OTUs) according to their sequence similarity (97%). A representative sequence for each OTU was selected with pick_rep_set.py script and used for taxonomic assignment using UCLUST and the Greengenes 13_8 database with assign_taxonomy_uclust.py. Sequences were aligned (align_seqs_pynast.py script) using PyNAST (Caporaso et al., 2010a) and filtered (filter_alignment.py script). A phylogenetic tree was subsequently produced with the make_phylogeny.py script. Singleton OTUs and OTUs whose representative sequences could not be aligned with PyNAST were removed.
After sample size standardization to 4,400 sequences/sample, OTU richness, alpha and beta-diversity metrics were calculated. The computed alpha diversity indexes were: Chao1, Shannon index, and Evenness. Two beta diversity metrics were computed: unweighted and weighted UniFrac distances. These were chosen because they use phylogenetic information and consequently yield results that are more useful (Lozupone and Knight, 2005; Hamady and Knight, 2009). In addition to the diversity metrics, prediction of functional composition of metagenome using the 16S rRNA gene as a marker was accomplished using PICRUSt (Langille et al., 2013). Predicted metagenome functions were performed using second-level KEGG pathways and can be found in the Supplemental Material (Tables S3, S4).
Statistical Analysis
Statistical analyses were performed using the software Minitab 18® and R v2.15.1 (R Core Team, 2013). Comparison of bacterial relative abundance was performed at the phylum and genus levels by a two-sample t-test with a confidence level of 95% in which the group variances were individually calculated. Results were considered significant at P ≤ 0.05 and were treated as trends when their associated P-values were between 0.05 and 0.10.
For the α diversity metrics, comparisons were performed between groups using a two-sample t-test with the same assumptions previously described. Beta diversity was accessed using the unweighted UniFrac distances matrices, which contained the dissimilarity values for each pairwise comparison. Analyses of sample groups were performed by permutational multivariate analysis of variance (PERMANOVA) with the significance being determined using 999 permutations.
RESULTS
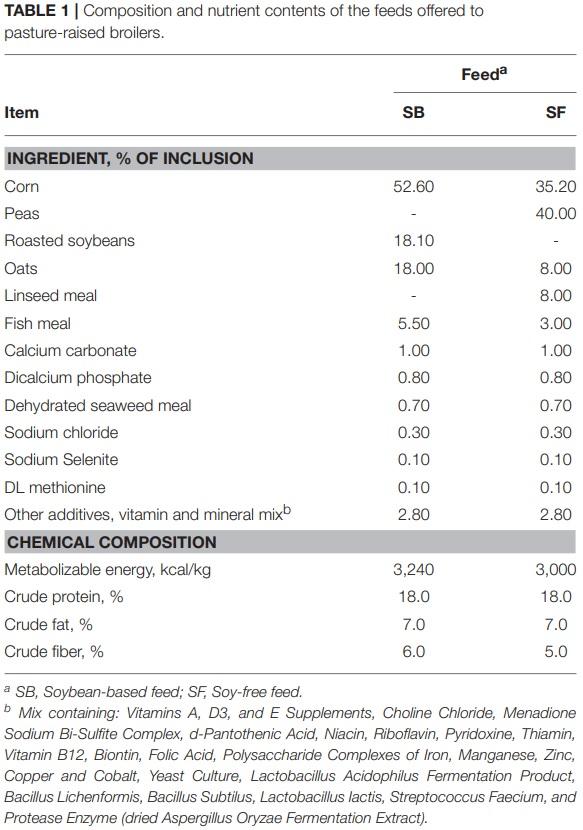
Only the main results are presented here. The interested reader may refer to the supplementary material (Tables S1–S4, and Figures S1–S3) for additional information. After all quality control steps, the samples that were used in the analysis yielded a total of 4,917,922 cleaned reads, resulting in an average of 33,229 reads per sample, with a range of 4,465 to 213,116 reads. As the birds were raised, microbial richness, diversity, and evenness remained relatively constant for the 3 fecal samples collected, regardless of diet (Table 2). However, the samples obtained when birds were processed tended to have a lower microbial richness with greater evenness, indicating a lower microbial abundance with a greater degree of similarity in the latter type of samples, regardless of the diet fed. When comparing the α-diversity metrices between diets, there were no significant differences in terms of richness (Chao1) in fecal samples from 4- and 7-week-old broilers; however, for day of processing (12-weekold) feces and all postharvest samples (i.e., cecal contents and WCR from the processing stage, and WCR on the final product) richness was significantly higher (P ≤ 0.05) in the SF diet. With regards to sample diversity (Shannon index), while there were significant differences based on diet for 3 sample types (7- week-old feces, 12-week-old feces, cecal contents), there was no consistent trend between diets as was observed with the richness estimates. Similarly, although some significant differences in sample evenness were observed between the SB and SF diets, no clear patterns were observed for this index.
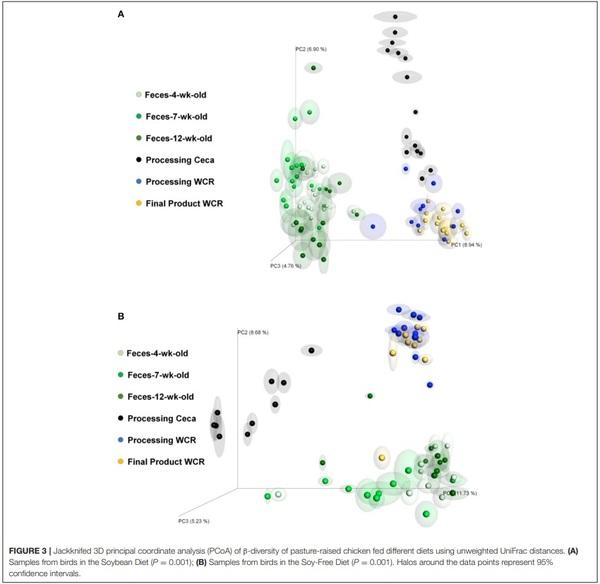
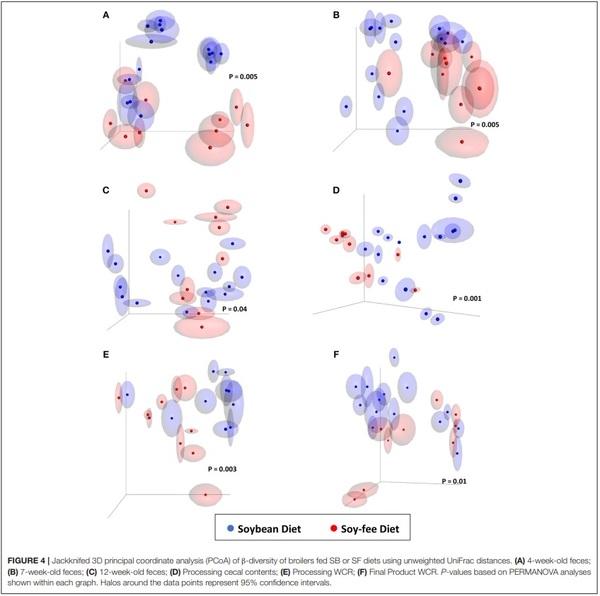
Jackknifed 3D principal coordinate analyses (PCoA) were performed to assess β-diversity differences between SB and SF for the different sample types (Figures 3, 4). For both diets, there was a clear pattern in which the fecal samples tended to be more similar to each other and therefore clustered together, apart from the other kind of samples. Similarly, samples from the ceca tended to be apart from the other sample types, whereas both WCR (from the processing stage and from the final product) tended to be more similar to each other (Figure 3). All of the 6 sample types exhibited significantly different (P ≤ 0.01) communities based on diet according to PERMANOVA analyses.
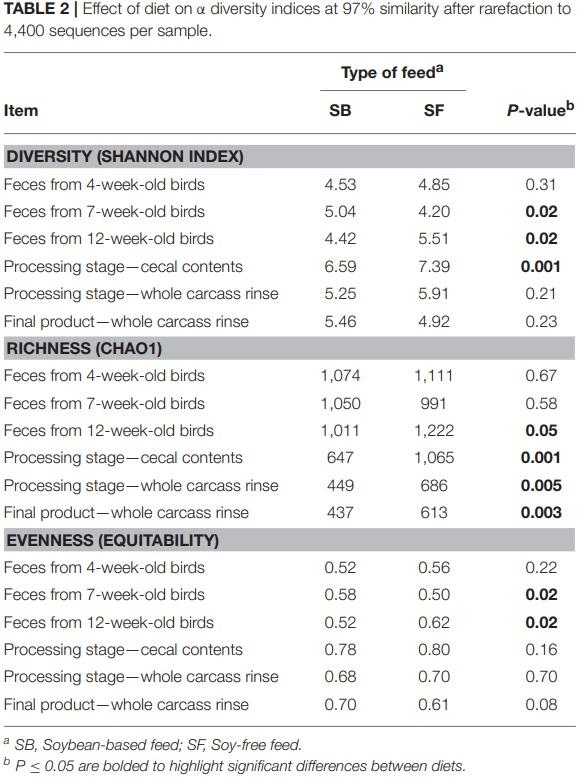
In addition to the effects on microbiome diversity along the farm-to-fork continuum, there were major diet-based differences observed. At the phyla level, no significant effect of diet (P ≥ 0.12) was detected in the feces collected when broilers were 4-weeksold; however, fecal samples collected at 7-weeks-old revealed a greater abundance (P = 0.0001) of Firmicutes, and lower abundance of Proteobacteria, Actinobacteria, and Bacteroidetes (P = 0.001, 0.002, and 0.02, respectively) for broilers fed the SF diet (Figure 1). Interestingly, these phyla-level relative abundance differences were all reversed by 12 weeks of age. During postharvest processing and in the final product, diet had a very limited effect on cecal or WCR microbiomes, with most phyla showing no significant difference between samples collected from broilers fed the SB or SF diets (Figure 2).
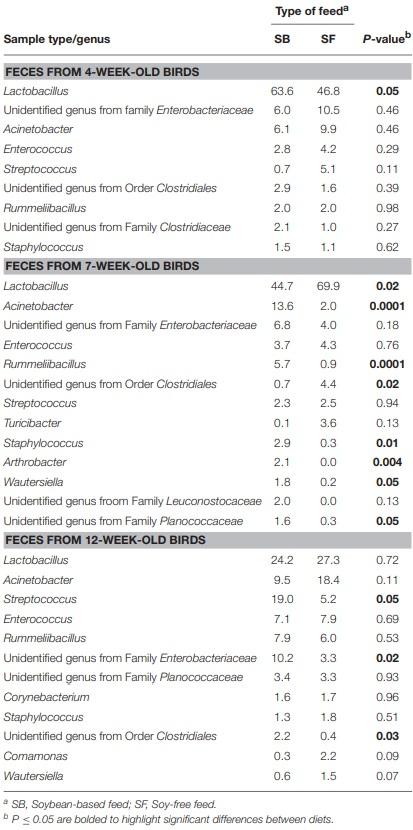
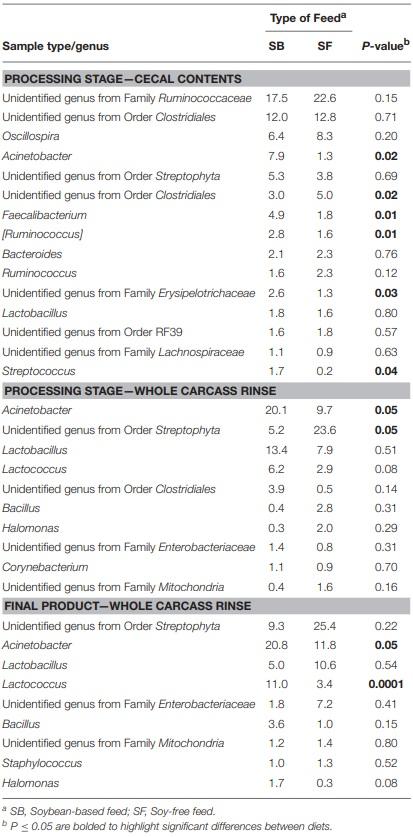
As seen at the phyla level, diet had the greatest impact at the genera-level preharvest taxa distribution in the 7-week-old broiler fecal samples (Table 3), with six dominant genera (≥ 1% of total OTUs in at least one diet:sample type group) significantly enriched in the SB diet, accounting for nearly 30% of all recovered OTUs in those samples. Conversely, only two genera were significantly enriched in the 7-week fecal samples of the broilers fed the SF diet, but they accounted for nearly 75% of all OTUs since Lactobacillus abundance (the most prevalent genera for all fecal samples tested) was significantly higher in SF. In no other preharvest fecal samples (4-week or 12-week) were any genera found to be significantly enriched for in the SF fed broilers. While there were other genera that were more abundant in the SBfed broiler fecal samples, none were significantly enriched for at more than a single sampling time. Postharvest microbiomes demonstrated similar trends (Table 4), with more genera being significantly enriched for in the SB-fed broilers, especially in the cecal samples where there were five genera significantly more abundant in the SB-fed broilers (∼20% of all OTUs) compared to only one genus more abundant in the SF-fed broilers (∼5% of all OTUs).
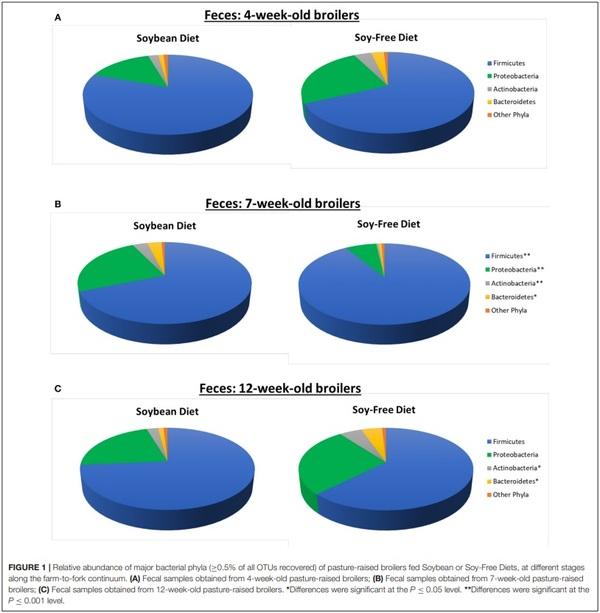
As previously mentioned, relative abundances of the main genera are shown in Tables 3, 4. For the first fecal samples (4-weeks-old broilers) the only genus found to be different across treatments was Lactobacillus, which had greater (P = 0.05) abundance in the group fed the SB diet. Conversely, for fecal samples collected 3 weeks later, the abundance of Lactobacillus was lower (P = 0.02) in the SB diet, however, the presence of Acinetobacter, Rummeliibacillus, Staphylococcus, and Arthrobacter were markedly greater (P ≤ 0.01) in the feces of birds consuming the SB diet. For the last fecal samples collected (12-weeks-old) the only significant changes detected were in Streptococcus, an unidentified genus from the family Enterobacteriaceae, and an unidentified genus from the order Clostridiales, which were all greater (P ≤ 0.05) in the group fed the SB diet. For cecal samples obtained in the processing stage, the genera Acinetobacter, Faecalibacterium, [Ruminococcus], and an unidentified genus from the family Erysipelotrichaceae were all greater (P ≤ 0.03) in the SB diet. Whole carcass rinses from the processing stage revealed a lower (P = 0.05) abundance of an unidentified genus of the order Streptophyta in the SB diet, but a greater abundance of Acinetobacter, which persisted in samples collected afterwards (whole carcass rinses of the final product); however, this latter type of sample also revealed a superior (P = 0.0001) abundance of Lactococcus in the SB diet, which had been found to be only a trend (P = 0.08) in the whole carcass rinses from the processing stage.
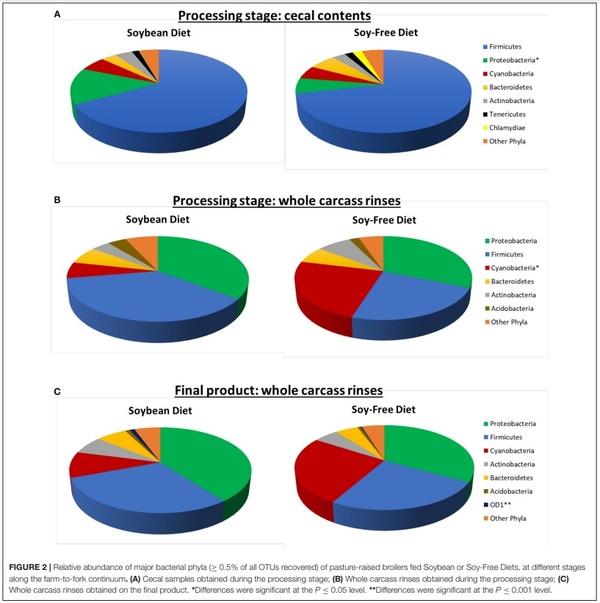
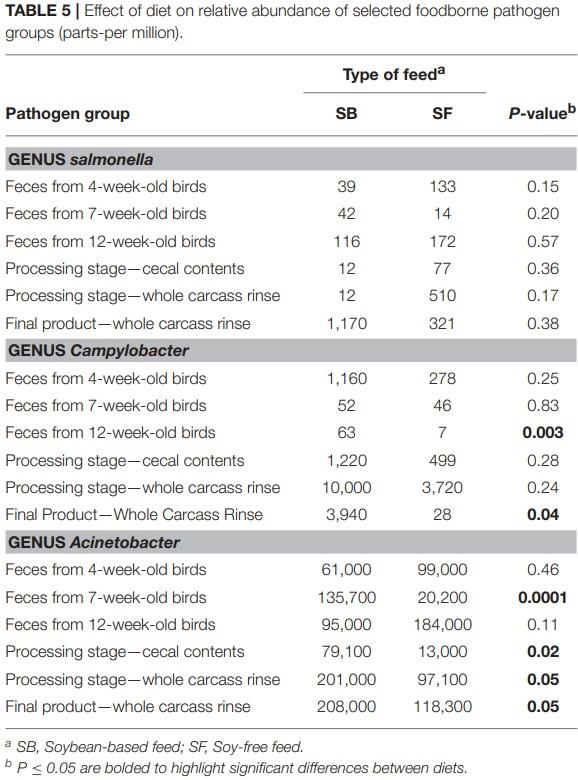
The presence of 2 important foodborne pathogens associated with poultry—Salmonella and Campylobacter—were quantified in all samples. In addition, one opportunistic microorganism group—Acinetobacter—which may act as pathogen under certain circumstances was also quantified (Table 5). No significant differences (P ≥ 0.15) were detected in the abundance of Salmonella in any of the types of samples. Although 2 types of samples showed numerically higher values of Acinetobacter in the SF diet (i.e., feces from 4 and 12-week-old birds), these differences were not significant (P ≥ 0.11). However, for all the other sample types analyzed in this study, abundances of Acinetobacter were greater (P ≤ 0.05) in birds fed the SB diet. Broilers fed the SF diet had consistently lower abundances of Campylobacter during their entire lifecycle. Such abundances were numerically inferior throughout all types of sample analyzed, and they were particularly important in the feces of 12-week-old broilers (P = 0.003) and in whole carcass rinses obtained from the final product (P = 0.04). In the former type of sample, abundance of Campylobacter was 9.5 times higher in the SB diet compared to the SF diet, whereas, in the latter type of sample this difference was more prominent: 142 times higher.
TABLE 3 | Relative abundance of major bacterial genera (≥ 1% of all OTUs recovered) for pasture-raised broilers fed soybean or soy-free diets at different stages along the farm-to-fork continuum: fecal samples from 4, 7, and 12-week-old broilers.
TABLE 4 | Relative abundance of major bacterial genera (≥ 1% of all OTUs recovered) for pasture-raised broilers fed soybean or soy-free diets at different stages along the farm-to-fork continuum: cecal contents and whole carcass rinses.
DISCUSSION
The effect of replacing feed ingredients on the gastrointestinal microbiome of chickens has been demonstrated. Díaz Carrasco et al. (2018) included tannins in the diet of growing broilers and detected a significant impact on cecal microbiota diversity: animals treated with tannins showed greater diversity than the untreated ones. In addition, significant changes in β-diversity were observed between those groups. In the present study, the replacement of soybeans with peas in SF likely resulted in different levels of tannin in the 2 tested diets, which may have contributed to the observed differences in α- and β-diversities (Smulikowska et al., 2001). Furthermore, the greater percentage of fat usually found in soybeans compared to peas, and the different amino acid profile between these two feed ingredients (National Research Council–NRC, 1994) likely played a role in the observed results. Interestingly, a combined assessment of α- and β-diversities show that the SF diet significantly affected the mature broiler GIT microbiome (12-week-old feces and cecal content samples from same day processing), potentially resulting in the different final product WCR microbiomes between diets (Table 2—Chao1 and Figure 4). These results support previous work that has shown that microbial populations in commercial/conventional postharvest environments can be directly affected by the microbiota present on farm directly prior to processing (Berghaus et al., 2013).
Several studies have demonstrated that the replacement and/or inclusion of ingredients in chicken diets generates important shifts in the major GIT phyla. Factors such as minerals (Tilocca et al., 2016), and the source of protein (Pineda-Quiroga et al., 2018) in the diet have been investigated and shown to significantly impact the GIT microbiome in commercial chickens. The replacement of soybeans with peas as the main source of protein in the SF diet (as shown in Table 1) resulted in diets with very different nutrient profiles. For instance, soybean has 2.2 times more of the essential amino acid methionine, and almost 14 times more fat than do peas (National Research Council–NRC, 1994). Such nutritional differences likely changed the composition of what was available to the birds’ microbial population and played an important role in the GIT microbiome shifts that were observed in the broilers.
Campylobacter is one of the leading causes of foodborne illness in humans (Cole et al., 2006; Svetoch and Stern, 2010; World Health Organization (WHO), 2018). In addition, virtually all adult broilers in U.S. poultry farms are colonized by Campylobacter spp. (Svetoch and Stern, 2010). In such scenario, the poultry industry needs intervention strategies to reduce Campylobacter levels both during the preharvest phase and in the final product. However, given that the use of antibiotics in human food production is finding increasing resistance (Danzeisen et al., 2011; Mathews, 2016), controlling Campylobacter levels through modification of dietary ingredients is a strategy that is in alignment with the current trends in the food production chain, and that is precisely what was achieved by using the SF diet. Supplementing broiler diets with protein sources other than soybean may provide a different amino acid pattern in the diet. Distinct dietary amino acid patterns can potentially reduce the prevalence of Campylobacter infection in chickens (Visscher et al., 2017, 2018). Additionally, several studies indicated that soybean meal-based diets increase crude mucin excretion in the intestinal tract of birds (Adedokun, 2007; Horn et al., 2009; Visscher et al., 2018). The presence of mucin appears to be essential for the survival and growth of C. jejuni in the poultry gut (Van Deun et al., 2008). Moreover, soybeans are rich in non-starch polysaccharides which are not completely digested in the small intestine and can be fermented by the intestinal microbiota (Malathi and Devegowda, 2001; Lan et al., 2007; Rehman et al., 2008), resulting in formation of short chain fatty acids (Awad et al., 2016). Consequently, by removing soybean as the main protein source in the chicken diet, mucin and short chain fatty acid levels were likely affected, which contributed to the lower Campylobacter abundances observed in broilers fed the SF diet.
Acinetobacter spp. are proving to be among the most problematic pathogens facing contemporary clinicians (Evans et al., 2017). It commonly causes soft tissue and urinary infections, as well as several nosocomial infections at levels that are increasingly being reported (Wong et al., 2017). Thus, the relatively high levels of this pathogen found throughout ourstudy (from 1.3 to 20.8% of all the observed OTUs, or 13,000 to 208,000 ppm) is of concern, particularly in the WCR samples. However, the SF diet alleviated this issue: Acinetobacter levels in WCR from the SF diet were practically half of what was found in the SB diet, as can be seen in Table 5.
The exact mechanism by which the SF diet reduced the presence of the pathogens Campylobacter and Acinetobacter demands further investigation. Thus, a replication of the present study in a modified, more controlled environment is recommended. However, the numerically lower abundance of Campylobacter in all sample types analyzed, and lower abundance of Acinetobacter in all of the post-harvest samples in the SF diet, indicates that the presence of soybeans in chicken diets can be an important factor contributing to a greater presence of both Campylobacter and Acinetobacter in chicken products. Therefore, our data suggest that replacing the SB with the SF diet in the production of pasture-raised chickens is a viable strategy for producers. Not only does such a dietary switch appeal to a niche of potential consumers, but it also can reduce the presence of important foodborne pathogens in the final product presented to consumers.
This article was originally published in Frontiers in Sustainable Food Systems 3:36. doi: 10.3389/fsufs.2019.00036. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (CC BY).











")