Introduction
The differences in broiler chicken growth systems between conventional environmentally controlled housing and on pasture include rearing environments, feed components and what are considered acceptable dietary additives. Broiler chickens raised in conventional rearing systems are supplemented with antimicrobial agents or growth promoters for the purposes of production enhancement as well as improved health attributes. However, pasture raised chickens, as an alternative bird rearing system, are grown on fresh grass, outdoor environments without most traditional growth promoters except for feed additives considered natural or organic such as prebiotics [1–3]. Conventional commercial breeds are not always suitable for the longer growth periods and environmental conditions prevalent for outdoor management systems. Although slow-growing broiler breeds such as naked neck chicken, require a longer growth period (as much as 12 weeks) compared to fast-growing one (7 weeks), slow-growing broiler do yield a better gait score, improved livability and nutritional differences in meat quality [4, 5].
However, issues such as occurrence of foodborne pathogens and diminished health status can be a chronic problem in some of the nonconventional systems [6]. Even in more conventional poultry production systems, emergence of multidrug resistant bacteria in poultry products and rearing system environments as well as the demands for high quality foods by consumers has accelerated the development of alternative feed amendments [2, 6]. Functional feed additives such as prebiotics have been examined in the past several decades as potential dietary additives to limit pathogenic bacteria establishment in humans and improve gut health in poultry and other food animals [7–10].
Prebiotics are not digestible by the host but commensal bacteria in the gut can metabolize them to produce short chain fatty acids (SCFAs) [8] and in some cases bacteriocins to inhibit the colonization of pathogenic bacteria in the gut as well as select for beneficial bacteria such as Lactobacillus and Bifidobacteria. Furthermore, since the complex ecosystem of chicken gastrointestinal tracts (GIT) plays an important role in nutrient utilization [11], growth development [12], detoxification [13], villi and crypt promotion [11], it is crucial to maintain microbial populations that support healthy host conditions [14]. Several commercial prebiotic type components have been generated from yeast cells including cell walls and fermentation products [15– 17]. These prebiotic type components not only have positive effects on animal and poultry productivity but also contribute to a healthy gut physiology along with the increased shift towards beneficial microorganisms [15–18].
Since prebiotics have a potential impact on gut health and are generally recognized as safe (GRAS) status, nonconventional food animal production systems can potentially use prebiotics to enhance productivity efficiency. While several reports have evaluated the effects of prebiotics on growth performance and meat quality [4, 9, 10, 19], the impacts of prebiotics on gut microbiota in nonconventional food animal production systems have not been fully explored [2]. Although a denaturing gradient gel electrophoresis (DGGE) technique has been widely utilized to investigate the microbiota differences between control and treatments, similar banding patterns on a gel make it difficult to interpret differences as well as the problems associated with the low DNA recovery rate from the gel for sequencing being imprecise for delineating the more subtle changes in microbiota [2, 20, 21].
High-throughput next generation sequencing (NGS) platforms based on 16S rRNA gene amplicons can serve as a phylogenetic markers to explore the more complex aspects of the microbiome in humans and animals [12, 22–26]. Amplicon sequencing using an Illumina MiSeq platform is rapidly increasing since the MiSeq possesses a lower cost per sequence compared to other platforms [27] and generates 7.5 Gb from 15 million 250-base paired-end reads within 3 days as well as longer sequences when using the MiSeq Reagent Kit v2 500 cycles (Illumina, San Diego, CA, USA).
Even though there have been several trials involving the naked neck breed of birds in pasture flocks, most of them have focused on the reduction of pathogens such as Salmonella and Campylobacter and growth performance [4, 9, 21, 28]. Investigation of the gut microbiota in naked neck chickens raised on pasture flock was initiated in the current study to focus on the implications of prebiotic roles on commensal microorganisms as a follow-up to an earlier study [21]. To the best of our knowledge, this is the first study to investigate pasture flock raised naked neck chicken cecal microbiota fed with yeast-based prebiotics using the Illumina MiSeq platform. In order to evaluate the effects of two commercial prebiotics Biolex® MB40 and Leiber® ExCel derived from yeast cell walls on chicken gut microbiota, naked neck chickens were grown up to 8 weeks on pasture flock fed with one of these prebiotics. Total DNA isolated from each cecum were utilized for sequencing using the Illumina MiSeq platform and quantitative insights into microbial ecology (QIIME) pipeline as a bioinformatics tool was adopted for sequencing data analysis and interpretation.
Materials and Methods
Chicken housing
A total of 147 naked neck chicks (Peterson Farms, Decatur, AR, USA) were randomly allocated to 3 pens (49 birds per each pen) with feed and water ad libitum for the duration of the 8 week experimental period. The birds and each pen were relocated within the pasture twice a week to supply fresh growing conditions. One pen served as the control (C) group fed with only genetically modified organism (GMO)-free feeds (Hiland Naturals, Killbuck, OH, USA) and the other two groups (T1 and T2) were fed with one of the respective yeast-based prebiotics with typical feed mixtures in the starter, grower and finisher rations. The feeds of both T1 and T2 groups were mixed with prebiotics Biolex® MB40 (0.2%, Leiber GmbH, Hafenstraße, Germany) derived from yeast cell (Saccharomyces cerevisiae) walls included beta-D-glucan and mannan-oligosaccharides (MOS) and Leiber® ExCel (0.2%, Leiber GmbH) which is similar to Biolex® MB40, respectively, and supplemented for the duration of the experimental period. The Institute Animal Care and Use Committee (IACUC) approval was exempted for this research because all birds were commercially raised in an off-campus facility and limited to microbiological evaluation.
Sample collection
At 8 weeks, a total of 45 birds (15 birds per group) were randomly chosen and humanely euthanized with CO2 gas. The extracted ceca were immediately transferred to sterile Whirl-Pak® bags (Nasco, Fort Atkinson, WI, USA) individually and stored -20°C until DNA extraction. The remainder of the birds were processed and the respective performance responses were described previously [21].
DNA extraction
A total of 200 mg of cecal contents from each bird was utilized for DNA isolation using a QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) with modifications to increase DNA concentration. The specific DNA extraction protocol was described previously [21]. Isolated DNA concentration was measured using a Qubit® 2.0 Fluorometer (Life Technology, Carlsbad, CA, USA) and diluted to 10 ng/μL.
Library preparation
A 10 ng of DNA aliquot isolated from each cecal content sample was utilized to construct a sequencing library targeting the V4 region of 16S rRNA following a previous report [29]. For library preparation, 40 samples from another independent study were combined with 45 samples to simultaneously sequence both sets of samples. In brief, individual DNA samples were amplified with dual-index primers via PCR and normalized amplicons using a SequalPrep™ Normalization kit (Life Technology) according to the manufacturer’s recommendation. Each sample possessed specific barcode sequences at the front and end of the PCR amplicon to discriminate among each other in the pooled library. A five microliter aliquot of each normalized sample was combined to generate 1 pooled library for further assays. Both library concentration and an exact product size were measured using a KAPA Library Quantification Kit (Kapa Biosystems, Woburn, MA, USA) through a quantitative PCR (qPCR, Eppendorf, Westbury, NY, USA) assay and an Agilent 2100 Bioanalyzer System (Agilent, Santa Clara, CA, USA), respectively. Based on the qPCR and bioanalyzer results, the pooled library was subsequently diluted to 4 nM prior to sequencing.
Sequencing via an Illumina MiSeq platform
A pooled library (20 nM) and a PhiX control v3 (20 nM) (Illumina) were mixed with 0.2 N fresh NaOH and HT1 buffer (Illumina) to produce the final concentration at 12 pM each. The resulting library was mixed with the PhiX control v3 (5%, v/v) (Illumina) and 600 uL loaded on a MiSeq® v2 (500 cycle) Reagent cartridge for sequencing. All sequencing procedures were monitored through the Illumina BaseSpace® website.
Sequencing data processing
Both demultiplexed R1 and R2 sequencing reads (approximately 250 bp in length) files were acquired from the Illumina BaseSpace® website and data processing were performed using a QIIME pipeline (version 1.9.0) [30]. The clustered sequences were utilized to construct Operational Taxonomic Units (OTUs) tables with 97% identity and representative sequences were classified into the respective taxonomical level from phylum to genus based on the Greengenes 16S rRNA gene database. Subsequently alpha diversity (rarefaction curve for OTUs, Chao1, and PD_Whole_Tree) and beta diversity using weighted and unweighted UniFrac distance among samples were generated within the QIIME 1.9.0 package.
Results and Discussion
Gastrointestinal tract microbiota analysis
The chicken ceca, as a fermentation chamber, not only play important roles such as polysaccharide digestion, water adsorption and urea recycling but also have the greatest gastrointestinal microbial populations that include an abundance of phylogenetic groups such as Clostridiales and Bacteroidetes [31, 32]. Recently, Sergeant et al. [26] reported that chicken ceca possess approximately 700 bacterial species based on 16S rRNA amplicon pyrosequencing.
In order to investigate chicken gut microbial populations, several approaches based on viable cells via cultural methods and molecular technologies such as DGGE have been adopted and utilized [21, 33, 34]. The DGGE based on DNA extracted from chicken cecal contents has been utilized as an alternative molecular-based technology over culture-dependent methods; however it does not necessarily represent overall microbiota due to limitations such as low bacterial discernment and insufficient diversity representation [20, 21, 35, 36, 37]. As an alternative approach, the qPCR assay based on 16S rRNA gene clones has been utilized to quantify complex microbiota in chicken ceca [38, 39] but the qPCR assay also has limitations such as PCR primers bias [40].
The NGS technology has been developed over past decade and decreasing costs per sequence has allowed for enhanced characterization and profiling of microbiota in complex ecosystems [27]. The Illumina MiSeq platform, bench top sequencer, based on 16S rRNA amplicons is widely utilized to generate 1.5 Gb per one day with 5 million 150-base paired-end reads [27]. Zhao et al. [41] evaluated the influence of different genotypes and gender on the corresponding chicken fecal gut microbiome based on the V4 regions of 16S rRNA using the Illumina MiSeq platform. A total of 68 out of 190 microbiome species were affected by gender and genotypes and 16 species were identified as Lactobacillus. In addition, Fadrosh et al. [22] utilized a similar dual-indexing approach used in the current study for multiplexed 16S rRNA amplicon sequencing based on the Illumna MiSeq platform.
Sequencing data analysis with QIIME
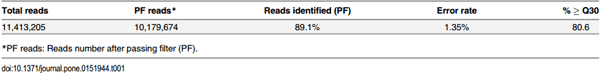
The Illumina MiSeq was performed using 45 independent samples to generate a total 11,413,205 raw sequence reads (Table 1). After passing the quality filter in an Illumina BaseSpace®, 10,179,674 (89.1%) an average of 236,737 reads (Table 1) were utilized for downstream analysis via the QIIME (1.9.0) pipeline [30]. The range of reads used in QIIME analysis was 89,350 to 281,996 and the information for each of the sample reads are illustrated in the S1 Table.
Sequencing data in this study exhibited a 1.35% error rate and 80.6% quality score, respectively (Table 1). Error rate is considered an important factor to evaluate the sequencing quality and associated with quality score (% ≥ Q30) reported by the instrument [29]. A low quality score can increase the error rate including nucleotide substitutions, insertions, deletions, and ambiguous base calls [29].
Microbial correlation among groups
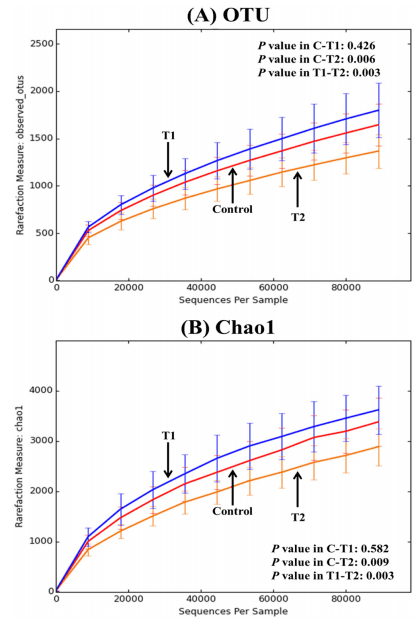
Both alpha and beta diversity were generated using the QIIME 1.9.0 package with a script core_diversity_analyses.py. For alpha diversity analysis, each rarefaction of average observed_OTUs and Chao1 per group is shown in Fig 1A and 1B, respectively. As shown in Fig 1A, T1 (Biolex® MB40) and control groups possessed similar unique OTU numbers, while T2 (Leiber® ExCel) exhibited a significantly lower specific OTU number compared to the other two groups. The Chao1 rarefaction plot (Fig 1B), estimating species richness, also displayed similar patterns to the observed_OTU rarefaction.
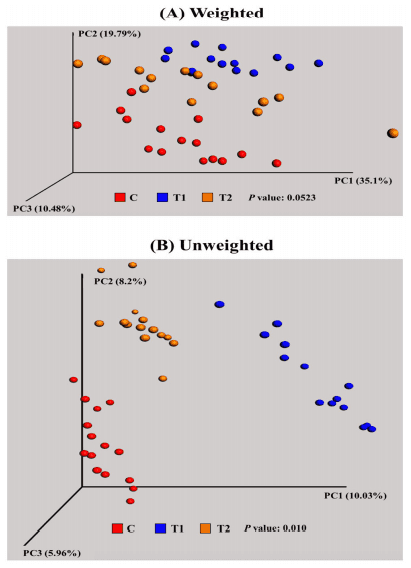
In the beta diversity analysis, both weighted and unweighted principal coordinated analysis (PCoA) UniFrac plots were generated using a total of 43 samples (Fig 2A and 2B). The PCoA plot served as a multivariate statistical method to indicate the phylogenetic distance between samples with 2 or 3 dimensional presentation diagrams. The weighted PCoA UniFrac plot exhibited the relative abundance of OTUs among group, while the unweighted PCoA UniFrac plot represented the phylogenetic distance based on the presence/absence of OTUs among samples. Fig 2A illustrates the weighted PCoA UniFrac plot and each dot (individual sample) in each group aligned in parallel on the PC1 (41.23%) axis, while each group is clustered distinctively in the unweighted PCoA UniFrac plot as shown in Fig 2B.
Taxonomic summary
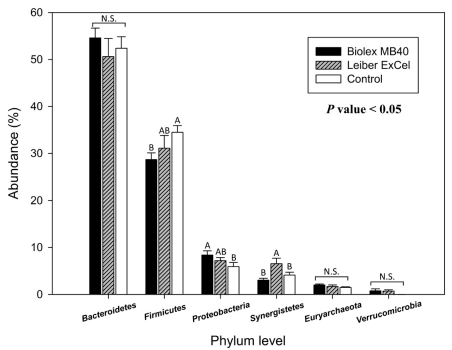
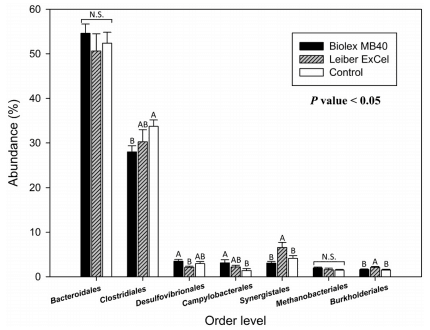
All bacterial group taxonomical summaries from phylum to genus levels in individual cecal contents and relative OTUs abundances are illustrated in S1 Fig and S2 Table. Both Figs 3 and 4 represent the top 6 and 7 bacterial groups at a phylum and order level, respectively. In the Fig 3, there were no apparent differences in Bacteroidetes and Euryarchaeota among groups, while the control group exhibited a significantly greater OTU abundance for Firmicutes (Fig 3). The T1 (Biolex® MB40) group revealed relatively high OTUs abundances in Proteobacteria and Cyanobacteria compared to the other two groups (T2; Leiber® ExCel and control) and Synergistetes OTUs was the highest in T2 (Leiber® ExCel) group.
Table 1. Sequencing data analysis
According to the previous reports by Ley et al. [42] and Mariat et al. [43], a decrease in the phylum Firmicutes/Bacteroidetes ratio is directly associated with a weight loss in humans and mice. In this study, there was no significant difference in the Firmicutes/Bacteroidetes ratio among groups (P > 0.05, data not shown) and this result is highly consistent with a previous report on these birds [21] which included the body weight responses of these same birds which served as the source for the cecal DNA samples used for sequencing in the current study. Although microbiota in chicken ceca evolves from birth to death, several papers have reported that Firmicutes remain the predominant bacteria in bird ceca raised in conventional rearing systems [12, 44–46]. However, Bacteroidetes do appear to be the predominant bacteria in chickens raised on pasture perhaps due to the different rearing systems [47], bird type [45] and dietary diversity dependent on exposure to sources such as consistent consumption of insects outdoors.
Fig 1. Rarefaction curves of alpha diversity among groups. (A) average observed_OTUs at 97% of similarity and (B) average Chao1; Control: normal feed, T1: normal feed with 0.2% Biolex® MB40 and T2: normal feed with 0.2% Leiber® ExCel.
Fig 2. Beta diversity analysis among groups. (A) weighted and (B) unweighted UniFrac PCoA plots of individual birds in each group. Individual sample was represented as spot with red (C; normal feed), blue (T1; normal feed with 0.2% Biolex® MB40), and orange (T2; normal feed with 0.2% Leiber® ExCel).
Fig 3. Comparison of top 6 cecal bacteria among groups in a phylum level. Blue, red, and green bars stand for T1 (normal feed with 0.2% Biolex® MB40), T2 (normal feed with 0.2% Leiber® ExCel), and control (C; normal feed), respectively. Capital letters on the top of bar within each phylum level indicate significant differences among groups (P < 0.05). N.S. (not significant).
In the top 7 bacterial groups at the order level, Bacteroidales and archaeal Methanobacteriales were found in chicken ceca [48], but both bacteria were not significantly different among the treatment groups in the current study (Fig 4). The control group did reveal a significant level of OTUs corresponding to Clostridiales, while the other three bacterial groups (Campylobacterales, Synergistales and Burkholderiales) exhibited lower OTUs abundances in the control group. Danzeisen et al. [45] compared the chicken cecal microbiome fed with antibiotics which have historically been used in the U.S. poultry industry [49] in order to demonstrate microbial changes impacted by age and treatments. Danzeisen et al. [45] also concluded Clostridiales is a predominant order in the Firmicutes phylum. In addition, Lu et al. [50] when evaluating the bacterial communities in ceca from mature broilers over time up to 49 days via 16S rDNA clone library reported that Clostridiaceae were detected as a predominant group in samples from all ages of birds.
Bacterial OTUs abundances in family and genus taxonomic level
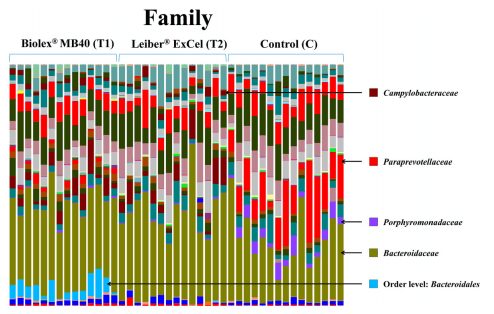
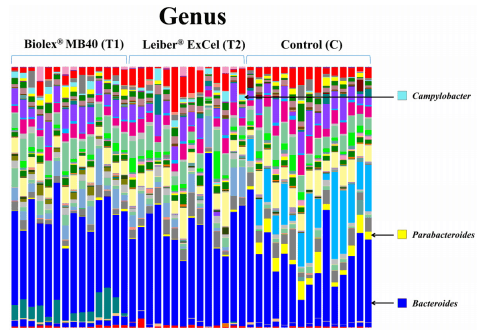
Figs 5 and 6 represent the relative distributions of OTUs in the cecal contents from each bird as a family and genus taxonomical level, respectively. As shown Fig 5, the control group exhibited distinct OTUs abundances at the family level compared to the other two treatment groups (T1: Biolex® MB40 and T2: Leiber® ExCel). Both families, Paraprevotellaceae and Porphyromonadaceae, were detected in all birds from the the control group with high amounts of OTUs and Porphyromonadaceae was further identified as genus Parabacteroides (Fig 5). The family Campylobacteraceae (Fig 5), genus Campylobacter (Fig 6), was present in all groups analyzed but the two treatment groups (T1: Biolex® MB40 and T2: Leiber® ExCel) exhibited significantly greater OTU abundance (Table 2). Interestingly, some bacteria from the order Bacteroidales OTUs (Fig 5) were detected only in the T1 (Biolex® MB4) group but could not be further assigned as a particular family and genus level.
Fig 4. Comparison of top 7 cecal bacteria among groups in an order level. Blue, red, and green bars stand for T1 (normal feed with 0.2% Biolex® MB40), T2 (normal feed with 0.2% Leiber® ExCel), and control (C; normal feed), respectively. Capital letters on the top of bar within each order level indicate significant differences among groups (P < 0.05). N.S. (not significant).
Fig 5. Overall cecal microbiota compositions of each sample with a family level. The 15 samples from left are T1 (normal feed with 0.2% Biolex® MB40), 15 samples in the middle are T2 (normal feed with 0.2% Leiber® ExCel) and next 15 samples are control (C; normal feed), respectively.
Fig 6. Overall cecal microbiota compositions of each sample with genus level. The 15 samples from left are T1 (normal feed with 0.2% Biolex® MB40), 15 samples in the middle are T2 (normal feed with 0.2% Leiber® ExCel) and next 15 samples are control (C; normal feed), respectively.
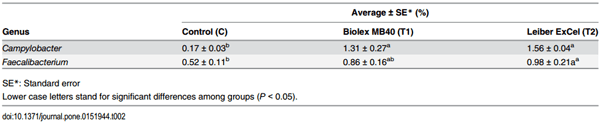
Table 2. Average of Campylobacter OTU abundance in a genus level among groups.
Effects of prebiotics on microbial populations
Although prebiotics based on yeast cell walls have been investigated their effects on gut microbial populations of chickens and applied to poultry industry in order to reduce foodborne pathogens, the exact mechanisms and functions of these prebiotics remain unclear and inconsistent results have been reported [51–53]. The MOS which is a main component of prebiotics used in this study has been known to not only enhance growth performance of birds [54] but also bind to the mannose-specific type-1 fimbriae receptor of pathogens to prevent colonization [55]. In addition, MOS may stimulate the growth of beneficial bacteria and enhance host immune responses [53]. In this study, there was a significant increase in genus Faecalibacterium OTUs, representing the phylum Firmicutes which are typically associated with health benefits as host commensal microorganisms [10] (Table 2).
Detection of Campylobacter
Campylobacter is a commensal bacterium in poultry but one of the most important foodborne pathogens to humans originating from poultry and poultry products. Throughout sequencing analysis, we sorted out Campylobacter prevalence at the genus level to evaluate the effects of two commercial prebiotics and all 45 birds possessed Campylobacter with a range of 4.91 to 0.01%. As an average of Campylobacter OTU abundance, there was no difference between the two prebiotics treated groups (T1: 1.43% and T2: 1.78%), while control group (C: 0.17%) exhibited significantly lower Campylobacter abundance compared to the other groups (Table 2; Fig 5). In order to confirm and support Campylobacter prevalence based on the sequencing results, quantitative PCR (qPCR) was performed according to the previous report [56], Similar to sequencing data, Campylobacter DNA copy numbers as a log value in both T1 (3.85 ± 0.17) and T2 (3.71 ± 0.17) groups were greater than the control (3.34 ± 0.16) group. In the previous report using these same samples as the current study, Park et al. [21] concluded that the control group exhibited a significant Campylobacter increase compared to the other two treatment groups using a conventional cultural method based on Campy-Cefex selective media (BD Biosciences, San Jose, CA, USA). However, we could not compare Campylobacter genus percentage of the sequencing data with the respective Campylobacter colony forming units (CFU) on selective media since sequencing data represented only the relative abundance within the entire sequenced bacterial population and 16S rRNA genes can have multiple copies on genomic DNA. In addition, although Campy-Cefex agar media have been widely utilized for the Campylobacter quantitation, several studies have reported its limitations on the detection ability [57, 58]. Line and Berrang [58] reported that Campy-Cefex media exhibited less inhibitory effects on the background bacteria in chicken carcass rinsates and Chon et al. [58] also noted similar results demonstrating that Campy-Cefex media exhibits a low isolation rate, accuracy and selectivity in the enriched chicken carcass rinsates.
Conclusions
Using high-throughput sequencing technologies, we were able to identify and delineate specific GIT microbiota patterns in long neck broilers fed yeast-based commercial prebiotics. We concluded that prebiotics additives derived from yeast cell walls altered cecal microbiota composition not only with changes in the phyla Firmicutes and Proteobacteria but also distinct changes in bacterial family and genus levels. Identifying specific microbial groups that tend to be predominant may help to better understand the interaction between host and GIT microbiota as well as microbiota changes when supplemented with food additives. Since the customized index primers utilized in this study for sequencing can generate a maximum of 384 different combinations, we could analyze over 300 samples in one reaction which offers advantages for larger studies utilizing more birds.
Acknowledgments
We would like to thank Franck G. Carbonero, Department of Food Science at the University of Arkansas, for use of the Illumina MiSeq instrument and Irene Hanning, Sandra Diaz, and Sean Pendleton, Department of Food Science and Technology at the University of Tennessee, for QIIME training.
Author Contributions
Conceived and designed the experiments: SHP SCR. Performed the experiments: SHP SIL. Analyzed the data: SHP. Contributed reagents/materials/analysis tools: SCR. Wrote the paper: SHP SCR.
This article was originally published in PLoS ONE 11(3): e0151944. doi:10.1371/journal.pone.0151944. This is an Open Access article distributed under the terms of the Creative Commons Attribution License.
References
1. AMS/USDA (2008) The national organic program. Agricultural Marketing Service-USDA, Washington, DC. Available: http://www.ams.usda.gov/about-ams/programs-offices/national-organic-program. Accessed 18 August 2015.
2. Park SH, Hanning I, Perrota A, Bench BJ, Alm E, Ricke SC. Modifying the gastrointestinal ecology in alternatively raised poultry and the potential for molecular and metabolomics assessment. Poult Sci. 2013; 92: 546–561. doi: 10.3382/ps.2012-02734 PMID: 23300323
3. Ricke SC. Potential of fructooligosaccharide prebiotics in alternative and nonconventional poultry production systems. Poult Sci. 2015; 94: 1411–1418. doi: 10.3382/ps/pev049 PMID: 25717086
4. Fanatico AC, Cavitt LC, Pillai PB, Emmert JL, Owens CM. Evaluation of slower-growing broiler genotypes grown with and without outdoor access: Meat quality. Poult Sci. 2005; 84: 1785–1790. PMID: 16463978
5. Fanatico AC, Owens CM, Emmert JL. Organic poultry production in the United States: Broilers. J Appl Poult Res. 2009; 18: 355–366.
6. Van Loo EJ, Alali W, Ricke SC. Food safety and organic meats. Annu Rev Food Sci Technol. 2012; 3: 203–225. doi: 10.1146/annurev-food-022811-101158 PMID: 22385165
7. Roberfroid MB. Introducing inulin-type fructans. Br J Nutr. 2005; 93: Suppl 1, S13–S25. PMID: 15877886
8. Józefiak D, Kaczmarek S, Rutkowski A. A note on the effects of selected prebiotics on the performance and ileal microbiota of broiler chickens. Anim Feed Sci Tech. 2008; 17: 392–397.
9. Pelicia K, Mendes AA, Saldanha ESPB, Pizzolante CC, Takahashi SE, Moreira J, et al. Use of prebiotics and probiotics of bacterial and yeast origin for free-range broiler chickens. Braz J Poult Sci. 2004; 6: 163–169.
10. Rastall RA, Gibson GR. Recent developments in prebiotics to selectively impact beneficial microbes and promote intestinal health. Curr Opin Biotechnol. 2015; 32: 42–46. doi: 10.1016/j.copbio.2014.11. 002 PMID: 25448231
11. Shakouri MD, Lji PA, Mikkelsen LL, Cowieson AJ. Intestinal function and gut microflora of broiler chickens as influenced by cereal grains and microbial enzyme supplementation. Anim Physiol Anim Nutr. 2009; 93: 647–658.
12. Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. 2012; 13: 89–99. doi: 10.1017/S1466252312000138 PMID: 22853945
13. Hai Y, Zhou T, Gong J, Young C, Su X, Li XZ, et al. Isolation of deoxynivalenol-transforming bacteria from the chicken intestines using the approach of PCR-DGGE guided microbial selection. BMC Microbiol. 2010; 10: 182. doi: 10.1186/1471-2180-10-182 PMID: 20576129
14. Kohl KD. Diversity and function of the avian gut microbiota. J Comp Physiol B. 2012; 182: 591–602. doi: 10.1007/s00360-012-0645-z PMID: 22246239
15. Ding G, Chang Y, Zhao L, Zhou Z, Ren L, Meng Q. Effect of Saccharomyces cerevisiae on alfalfa nutrient degradation characteristics and rumen microbial populations of steers fed diets with different concentrate-to-forage ratios. J Anim Sci Biotech. 2014; 5: 24.
16. Santin E, Maiorka A, Macari M. Performance and intestinal mucosa development of broilers chickens fed diets containing Saccharomyces cerevisiaes cell wall. J Appl Poult Res. 2001; 10: 236–244.
17. Zhang AW, Lee BD, Lee SK, Lee KW, An GH, Song KB, et al. Effects of yeast (Saccharomyces cerevisiae) cell components on growth performance, meat quality, and ileal mucosa development of broiler chickens. Poult Sci. 2005; 84: 1015–1021. PMID: 16050118
18. Morales-López R, Auclair E, García F, Esteve-Garcia E, Brufau J. Use of yeast cell walls; β-1, 3/1, 6- glucans; and mannoproteins in broiler chicken diets. Poult Sci. 2009; 88: 601–607. doi: 10.3382/ps. 2008-00298 PMID: 19211531
19. Hanning I, Clement A, Owens C. Park SH, Pendleton S, Scott EE, et al. Assessment of production performance in 2 breeds of broilers fed prebiotics as feed additives. Poult Sci. 2012; 91: 3295–3299. doi: 10.3382/ps.2012-02557 PMID: 23155043
20. Park SH, Hanning I, Gilbert W, Munro M, Devareddy L, Ricke SC. Feeding mice aged and fresh blackberries powder supplements result in shifts in the gastrointestinal microflora. Food Biosci. 2013; 1: 66– 72.
21. Park SH, Gibson KE, Almeida G, Ricke SC Assessment of gastrointestinal microflora in pasture raised chickens fed two commercial prebiotics. J Prob Health. 2014; 2: 122.
22. Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014; 2: 6. doi: 10.1186/2049-2618-2-6 PMID: 24558975
23. Mojd Shaufi MA, Sieo CC, Chong CW, Gan HM, Ho YW. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015; 7:4. doi: 10.1186/ s13099-015-0051-7 PMID: 25806087
24. Oakley BB, Morales CA, Line JE, Seal BS, Hiett KL. Application of high-throughput sequencing to measure the performance of commonly used selective cultivation methods for the foodborne pathogen Campylobacter. FEMS Microbiol Ecol. 2012; 79: 327–336. doi: 10.1111/j.1574-6941.2011.01219.x PMID: 22092388
25. Oakley BB, Morales CA, Line J, Berrang ME, Meinersmann RJ, Tillman GE, et al. The poultry-associated microbiome: network analysis and farm-to-fork characterizations. PLoS One. 2013; 8: e57190. doi: 10.1371/journal.pone.0057190 PMID: 23468931
26. Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One. 2014; 9: e91941. doi: 10. 1371/journal.pone.0091941 PMID: 24657972
27. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME J. 2012; 6: 1621– 1624. doi: 10.1038/ismej.2012.8 PMID: 22402401
28. Sirri F, Castellini C, Bianchi M, Petracci M, Meluzzi A, Franchini A. Effect of fast-, medium- and slowgrowing strains on meat quality of chickens reared under the organic farming method. Animal. 2010; 5: 312–319.
29. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013; 79: 5112–5120. doi: 10.1128/AEM.01043-13 PMID: 23793624
30. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010; 7: 335–336. doi: 10. 1038/nmeth.f.303 PMID: 20383131
31. Deusch S, Tilocca B, Camarinha-Silva A, Seifert J. News in livestock research—use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Comput Struct Biotechnol J. 2015; 13: 55–63. PMID: 26900430
32. Meng H, Zhang Y, Zhao L, Zhao W, He C, Zhai Z et al. Body weight selection affects quantitative genetic correlated responses in gut microbiota. PLoS One. 2014; 9: e89862. doi: 10.1371/journal. pone.0089862 PMID: 24608294
33. Ricke SC, Pillai SD. Conventional and molecular methods for understanding probiotic bacteria functionality in gastrointestinal tracts. Crit Rev Microbiol. 1999; 25: 19–38. PMID: 10342098
34. Salanitro J, Fairchilds I, Zgornicki Y. Isolation, culture characteristics, and identification of anaerobic bacteria from the chicken cecum. Appl Microbiol. 1974; 27: 678–687. PMID: 4596749
35. Stanley D, Hughes RJ, Moore RJ. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl Microbiol Biotechnol. 2014; 98: 4301–4310. doi: 10.1007/s00253-014- 5646-2 PMID: 24643736
36. Diaz-Sanchez S, Hanning I, Pendleton S, D’Souza D. Next-generation sequencing: the future of molecular genetics in poultry production and food safety. Poult Sci. 2013; 92: 562–572. doi: 10.3382/ps. 2012-02741 PMID: 23300324
37. Zoetendal EG, Collier CT, Koike S, Mackie RI, Gaskin HR. Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr. 2004; 134: 465–472. PMID: 14747690
38. Tillman GE, Haas GJ, Wise MG, Oakley B, Smith MA, Siragusa GR. Chicken intestine microbiota following the administration of lupulone, a hop-based antimicrobial. FEMS Microbiol Ecol. 2011; 77: 395– 403. doi: 10.1111/j.1574-6941.2011.01119.x PMID: 21517917
39. Wise MG, Siragusa GR. Quantitative analysis of the intestinal bacterial community in one- to threeweek-old commercially reared broiler chickens fed conventional or antibiotic-free vegetable-based diets. J Appl Microbiol. 2007; 102: 1138–1149. PMID: 17381758
40. Suzuki MT, Giovannoni SJ. Bias caused by template annealing in the amplification of mixture of 16S rRNA genes by PCR. Appl Environ Microbiol. 1996; 62: 625–630. PMID: 8593063
41. Zhao L, Wang G, Siegel P, Chuan H, Wang H, Zhao W, et al Quantitative genetic background of the hos influences gut microbiomes in chickens. Sci Rep. 2013; 3:1163. doi: 10.1038/srep01163 PMID: 23362462
42. Ley RE, Bäcked F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. PNAS. 2005; 102: 11070–11075. PMID: 16033867
43. Mariat D, Firmesse O, Levenez F, Guimaraes VD, Sokol H, Doré J, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009; 9: 123. doi: 10.1186/1471- 2180-9-123 PMID: 19508720
44. Zhu XY, Zhong T, Pandya Y, Joerger RD. 16S rRNA-based analysis of microbiota from the cecum of broiler chickens. Appl Environ Microbiol. 2002; 68: 124–127. PMID: 11772618
45. Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulation of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One. 2011; 11: e27949.
46. Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One. 2008; 8: e2945.
47. Gabriel I, Lessire M, Mallet S, Guillot JF. Microflora of the digestive tract: critical factors and consequences for poultry. World’s Poult Sci J. 2006; 62: 499–511.
48. Saengkerdsub S, Herrera P, Woodward CL, Anderson RC, Nisbet DJ, Ricke SC. Identification and quantification of methanogenic archaea in adult chicken ceca. Appl Environ Microbiol. 2007; 73: 353– 356. PMID: 17085694
49. Jones FT, Ricke SC. Observation on the history of the development of antimicrobials and their use in poultry feeds. Poult Sci. 2003; 82: 613–617. PMID: 12710481
50. Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl Environ Microbiol. 2003; 69: 6816–6824. PMID: 14602645
51. Stanley VG, Gray C, Daley M, Krueger WF, Sefton AE. An alternative to antibiotic-based drugs in feed for enhancing performance of broilers grown on Eimeria spp.- infected litter. Poult Sci. 2004; 83:39–44. PMID: 14761082
52. Zhang AW, Lee BD, Lee SK, Lee KW, An GH, Song KB, et al. Effects of yeast (Saccharomyces cerevisiae) cell components on growth performance, meat quality, and ileal mucosa development of broiler chicks. Poult Sci. 2005; 84:1015–1021. PMID: 16050118
53. Gao J, Zhang HJ, Yu SH, Wu SG, Yoon I, Quigley J, et al. Effects of yeast culture in broiler diets on performance and immunomodulatory functions. Poult Sci. 2008; 87: 1377–1384. doi: 10.3382/ps.2007- 00418 PMID: 18577619
54. Hooge DM. Meta-analysis of broiler chicken pen trials evaluating dietary mannan oligosaccharide, 1993–2003. Int J Poult Sci. 2004; 3: 163–174.
55. Newman K. Mannan-oligosaccharides: natural polymers with significant impact on the gastrointestinal microflora and the immune system; Lyons TP, Jacques KA (Eds.), Biotechnology in the Feed Industry, Proceedings of Alltech’s 10th annual symposium, Nottingham University Press, Nottingham, UK. 1994; 167–174.
56. Park SH, Hanning I, Jarquin R, Moore P, Donoghue DJ, Donoghue AM, et al. Multiplex PCR assay for the detection and quantification of Campylobacter spp., Escherichia coli O157:H7, and Salmonella serotypes in water samples. FEMS Microbiol Lett. 2013; 316: 7–15.
57. Chon JW, Hyeon JY, Park JH, Song KY, Seo KH. Comparison of 2 types of broths and 3 selective agars for the detection of Campylobacter species in whole-chicken carcass-rinse samples. Poult Sci. 2012; 59: 2382–2385.
58. Line JE, Berrang ME. Comparison of two types of plating media for detection and enumeration of Campylobacter from poultry samples. Int J Poult Sci. 2005; 4: 81–84.










.jpg&w=3840&q=75)