Classification of infectious bursal disease virus into genogroups
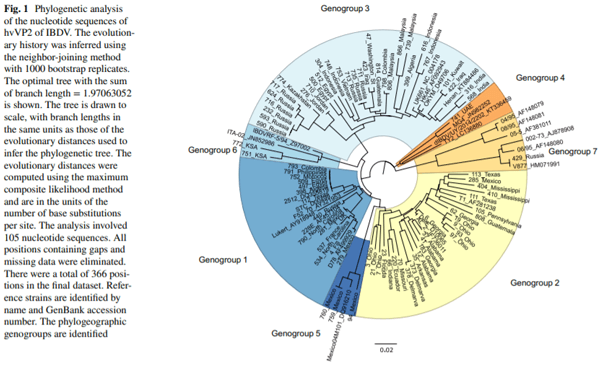
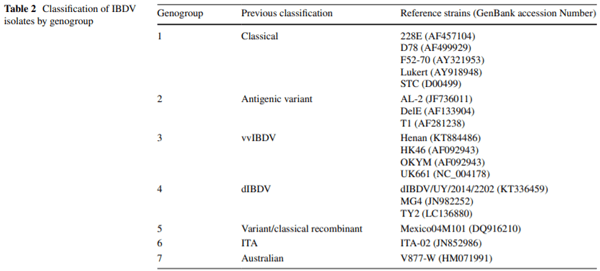
Infectious bursal disease virus (IBDV) causes infectious bursal disease (IBD), an immunosuppressive disease of poultry. The current classifcation scheme of IBDV is confusing because it is based on antigenic types (variant and classical) as well as pathotypes. Many of the amino acid changes diferentiating these various classifcations are found in a hypervariable region of the capsid protein VP2 (hvVP2), the major host protective antigen. Data from this study were used to propose a new classifcation scheme for IBDV based solely on genogroups identifed from phylogenetic analysis of the hvVP2 of strains worldwide. Seven major genogroups were identifed, some of which are geographically restricted and others that have global dispersion, such as genogroup 1. Genogroup 2 viruses are predominately distributed in North America, while genogroup 3 viruses are most often identifed on other continents. Additionally, we have identifed a population of genogroup 3 vvIBDV isolates that have an amino acid change from alanine to threonine at position 222 while maintaining other residues conserved in this genogroup (I242, I256 and I294). A222T is an important mutation because amino acid 222 is located in the frst of four surface loops of hvVP2. A similar shift from proline to threonine at 222 is believed to play a role in the signifcant antigenic change of the genogroup 2 IBDV strains, suggesting that antigenic drift may be occurring in genogroup 3, possibly in response to antigenic pressure from vaccination.

1. Dobos P, Hill B, Hallett R, Kells D, Becht H, Teninges D (1979) Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J Virol 32:593–605
2. Jackwood DJ, Sommer-Wagner SE (2011) Amino acids contributing to anitgenic drift in the infectious bursal disease birnavirus (IBDV). Virology 409:33–37
3. Jackwood DJ, Sreedevi B, LeFever LJ, Sommer-Wagner SE (2008) Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity. Virology 377:110–116
4. Benton WJ, Cover MS, Rosenberger JK, Lake RS (1967) Physiochemical properties of the infectious bursal agent. Avian Dis 11:438–445
5. Mandeville WFI, Cook FK, Jackwood DJ (2000) Heat lability of fve strains of infectious bursal disease virus. Poult Sci 79:838–842
6. Rautenschlein S, Yeh H, Njenga MK, Sharma JM (2002) Role of intrabursal T cells in infectious bursal disease virus (IBDV) infection: T cells promote viral clearance but delay follicular recovery. Arch Virol 147:285–304
7. Saif YM (1991) Immunosuppression induced by infectious bursal disease virus. Vet Immunol Immunopathol 30:45–50
8. Zachar T, Popowich S, Goodhope B, Knezacek T, Ojkic D, Willson P, Ahmed KA, Gomis S (2016) A 5-year study of the incidence and economic impact of variant infectious bursal disease viruses on broiler production in Saskatchewan, Canada. Can J Vet Res 80:255–261
9. Kurukulsuriya S, Ahmed KA, Ojkic D, Gunawardana T, Gupta A, Goonewardene K, Karunaratne R, Popowich S, Willson P, Tikoo SK, Gomis S (2016) Circulating strains of variant infectious bursal disease virus may pose a challenge for antibiotic-free chicken farming in Canada. Res Vet Sci 108:54–59
10. Van Den Berg TP, Morales D, Eterradossi N, Rivallan G, Toquin D, Raue R, Zierenberg K, Zhang MF, Zhu YP, Wang CQ, Zheng HJ, Wang X, Chen GC, Lim BL, Muller H (2004) Assessment of genetic, antigenic and pathotypic criteria for the characterization of IBDV strains. Avian Pathol 33:470–476
11. Bayliss CD, Spies U, Shaw K, Peters RW, Papageorgiou A, Muller H, Boursnell MEG (1990) A comparison of the sequences of segment A of four infectious bursal disease virus strains and identifcation of a variable region in VP2. J Gen Virol 71:1303–1312
12. Brandt M, Yao K, Liu M, Heckert RA, Vakharia VN (2001) Molecular determinants of virulence, cell trophism, and pathogenic phenotype of infectious bursal disease virus. J Virol 75:11974–11982
13. Eterradossi N, Arnauld C, Toquin D, Rivallan G (1998) Critical amino acid changes in VP2 variable domain are associated with typical and atypical antigenicity in very virulent infectious bursal disease viruses. Arch Virol 143:1627–1636
14. Eterradossi N, Toquin D, Rivallan G, Guittet M (1997) Modifed activity of a VP2-located neutralizing epitope on various vaccine, pathogenic and hypervirulent strains of infectious bursal disease virus. Arch Virol 142:255–270
15. Coulibaly F, Chevalier C, Delmas B, Rey F (2010) Crystal structure of an aquabirnavirus particle: insights into antigenic diversity and virulence determinism. J Virol 84:1792–1799
16. Coulibaly F, Chevalier C, Gutsche I, Pous J, Navaza J, Bressanelli S, Delmas B, Rey FA (2005) The Birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120:761–772
17. Letzel T, Coulibaly F, Rey FA, Delmas B, Jagt E, van Loon A, Mundt E (2007) Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus. J Virol 81:12827–12835
18. Islam MR, Rahman S, Noor M, Chowdhury EH, Müller H (2012) Diferentiation of infectious bursal disease virus (IBDV) genome segment B of very virulent and classical lineage by RTPCR amplifcation and restriction enzyme analysis. Arch Virol 157:333–336
19. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
20. Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
21. Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
22. Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101:11030–11035
23. Zuckerkandl E, Pauling L (1965) Evolutionary divergence and convergence in proteins. Evol Genes Proteins 97:97–166
24. Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
25. Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
26. Hernández M, Tomás G, Marandino A, Iraola G, Maya L, Mattion N, Hernández D, Villegas P, Banda A, Panzera Y, Prez R (2015) Genetic characterization of South American infectious bursal disease virus reveals the existence of a distinct worldwide-spread genetic lineage. Avian Pathol 44:212–221
27. Jackwood DJ (2012) Molecular epidemiologic evidence of homologous recombination in infectious bursal disease viruses. Avian Dis 56:574–577
28. Lupini C, Giovanardi D, Pesente P, Bonci M, Felice V, Rossi G, Morandini E, Cecchinato M, Catelli E (2016) A molecular epidemiology study based on VP2 gene sequences reveals that a new genotype of infectious bursal disease virus is dominantly prevalent in Italy. Avian Pathol 45:458–464
29. Shcherbakova LO, Lomakin AI, Borisov AV, Drygin VV, Gusev AA (1998) Comparative analysis of the VP2 variable region of the gene from infectious bursal disease virus isolates. Mol Gen Mikrobiol Virusol 1:35–40
30. Le Nouen C, Rivallan G, Toquin D, Eterradossi N (2005) Signifcance of the genetic relationships deduced from partial nucleotide sequencing of infectious bursal disease virus genome segments A or B. Arch Virol 150(2):313–325
31. Petkov D, Linnemann E, Kapczynski DR, Sellers HS (2007) Full-length sequence analysis of four IBDV strains with diferent pathogenicities. Virus Genes 34:315–326
32. Blake S, Ma JY, Caporale DA, Jairath S, Nicholson BL (2001) Phylogenetic relationships of aquatic birnaviruses based on deduced amino acid sequences of genome segment A cDNA. Dis Aquat Organ 45:89–102
33. Escafre O, Le Nouen C, Amelot M, Ambroggio X, Ogden KM, Guionie O, Toquin D, Muller H, Islam MR, Eterradossi N (2013) Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. J Virol 87:2767–2780
34. Van Den Berg TP (2000) Acute infectious bursal disease in poultry: a review. Avian Pathol 29:175–194
35. Jackwood DJ, Stoute ST (2013) Molecular evidence for a geographically restricted population of infectious bursal disease viruses. Avian Dis 57:57–64
36. Yamazaki K, Ohta H, Kawai T, Yamaguchi T, Obi T, Takase K (2017) Characterization of variant infectious bursal disease virus from a broiler farm in Japan using immunized sentinel chickens. J Vet Med Sci 79:175–183
37. Banda A, Villegas P, El-Attrache J (2003) Molecular characterization of infectious bursal disease virus from commercial poultry in the United States and Latin America. Avian Dis 47:87–95
38. Jackwood DJ, Sommer SE, Knoblich HV (2001) Amino acid comparison of infectious bursal disease viruses placed in the same or diferent molecular groups by RT/PCR-RFLP. Avian Dis 45:330–339
39. Ignjatovic J, Sapats S (2002) Confrmation of the existence of two distinct genetic groups of infectious bursal disease virus in Australia. Aust Vet J 80:689–694
40. Proftt JM, Bastin DA, Lehrbach PR (1999) Sequence analysis of Australian infectious bursal disease viruses. Aust Vet J 77:186–188
41. Brown MD, Green P, Skinner MA (1994) VP2 sequences of recent European ‘very virulent’ isolates of infectious bursal disease virus are closely related to each other but are distinct from those of ‘classical’ strains. J Gen Virol 75:675–680
42. Parede L, Sapats SI, Gould G, Rudd MF, Lowther S, Ignjatovic J (2003) Characterization of infectious bursal disease virus isolates from Indonesia indicates the existence of very virulent strains with unique genetic changes. Avian Pathol 32:511–518
43. Fernandes MJ, Simoni IC, Vogel MG, Harakava R, Rivas EB, Oliveira MB, Kanashiro AM, Tessari EN, Gama NM, Arns CW (2009) Molecular characterization of Brazilian infectious bursal disease virus isolated from 1997 to 2005. Avian Dis 53:449–454
44. Heine HG, Haritou M, Failla P, Fahey K, Azad AA (1991) Sequence analysis and expression of the host-protective immunogen VP2 of a variant strain of infectious bursal disease virus which can circumvent vaccination with standard type I strains. J Gen Virol 72:1835–1843
45. Jackwood DJ, Sommer-Wagner SE (2007) Genetic characteristics of infectious bursal disease viruses from four continents. Virology 365:369–375
46. Kasanga CJ, Yamaguchi T, Wambura PN, Maeda-Machang’u AD, Ohya K, Fukushi H (2007) Molecular characterization of infectious bursal disease virus (IBDV): diversity of very virulent IBDV in Tanzania. Arch Virol 152:783–790
47. Adamu J, Owoade AA, Abdu PA, Kazeem HM, Fatihu MY (2013) Characterization of feld and vaccine infectious bursal disease viruses from Nigeria revealing possible virulence and regional markers in the VP2 minor hydrophilic peaks. Avian Pathol 42:420–433
48. Nwagbo IO, Shittu I, Nwosuh CI, Ezeifeka GO, Odibo FJ, Michel LO, Jackwood DJ (2016) Molecular characterization of feld infectious bursal disease virus isolates from Nigeria. Vet World 9:1420–1428
49. Negash T, Gelaye E, Petersen H, Grummer B, Rautenschlein S (2012) Molecular evidence of very virulent infectious bursal disease viruses in chickens in Ethiopia. Avian Dis 56:605–610
50. Hoque MM, Omar AR, Chong LK, Hair-Bejo M, Aini I (2001) Pathogenicity of SspI-positive infectious bursal disease virus and molecular characterization of the VP2 hypervariable region. Avian Pathol 30:369–380
51. Islam MR, Zierenberg K, Muller H (2001) The genome segment B encoding the RNA-dependent RNA polymerase protein VP1 of very virulent infectious bursal disease virus (IBDV) is phylogenetically distinct from that of all other IBDV strains. Arch Virol 146:2481–2492
52. Alfonso-Morales A, Rios L, Martinez-Perez O, Dolz R, Valle R, Perera CL, Bertran K, Frias MT, Ganges L, Diaz de Arce H, Majo N, Nunez JI, Perez LJ (2015) Evaluation of a phylogenetic marker based on genomic segment B of infectious bursal disease virus: facilitating a feasible incorporation of this segment to the molecular epidemiology studies for this viral agent. PLoS One 10:e0125853
53. Gao L, Li K, Qi X, Gao H, Gao Y, Qin L, Wang Y, Shen N, Kong X, Wang X (2014) Triplet amino acids located at positions 145/146/147 of the RNA polymerase of very virulent infectious bursal disease virus contribute to viral virulence. J Gen Virol 95:888–897
54. Jackwood DJ, Crossley BM, Stoute ST, Sommer-Wagner SE, Woolcock PR, Charlton BR (2012) Diversity of genome segment B from infectious bursal disease viruses in the United States. Avian Dis 56:165–172
55. Owoade AA, Mulders MN, Kohnen J, Ammerlaan W, Muller CP (2004) High sequence diversity in infectious bursal disease virus serotype 1 in poultry and turkey suggests West-African origin of very virulent strains. Arch Virol 149:653–672
56. Jackwood DJ (2013) Multivalent virus-like-particle vaccine protects against classic and variant infectious bursal disease viruses. Avian Dis 57:41–50









.jpg&w=3840&q=75)