Introduction
The gastrointestinal (GI) tract of humans and animals is populated with a diverse group of microbes known as the microbiota that include bacteria, fungi, archaea, protists, and viruses, with bacteria being the most predominant [1, 2]. The bacterial microbiota is well known to be critically involved in host physiology and immune development [1, 2]; however, the role of the fungal community, known as the mycobiota, that plays in health and diseases is less studied and understood. Recent studies have suggested that a healthy intestinal mycobiota appears to be important for maintaining host homeostasis, modulating host immune responses, and competitive exclusion of pathogens [3–5]. For example, colonization of C. albicans protects mice against infections of virulent fungi and bacteria by stimulating the expansion of Th17 cells, activating neutrophils, and thus enhancing host defense against extracellular pathogens in mice [6, 7]. Alterations in the intestinal mycobiota are also linked to exaggerated inflammation in diseases such as human inflammatory bowel disease (IBD) [8, 9], allergic airway diseases [10, 11], and colorectal cancer [12]. The interplay between the intestinal mycobiota and microbiota is critical for intestinal homeostasis [4]. Studies have shown disease-specific bacteria-fungi networks [8, 13], highlighting the significance of the fungal community in host health and underscoring a need for further investigation of the mycobiome.
Little is known about the intestinal mycobiota in poultry. We recently revealed the mycobiota in the upper GI tract to be more diverse than the mycobiota in the lower GI tract of chickens [14]. Unlike the intestinal bacterial microbiota, which appears to become stabilized between d 21–28, the cecal mycobiota remains unstable beyond d 28 [14]. A study of the turkey ileal mycobiota revealed a similar kinetic trend [15]. Furthermore, alternations in the ileal mycobiome are significantly correlated with the bacterial changes in response to a lowdose antibiotic and probiotics [15]. However, the involvement of the intestinal mycobiota in poultry diseases has not been studied to date. The intestinal mycobiotamicrobiota interplay in the context of a disease remains unknown
Necrotic enteritis (NE), caused by pathogenic Grampositive bacterium C. perfringens, is one of the most common and economically devastating enteric diseases in poultry [16]. NE-induced disruption of the intestinal microbiota is well-documented [17]. Although the changes in the microbiota diversity vary among studies, the intestinal microbiota in NE chickens is generally characterized by an overgrowth of C. perfringens and Escherichia/Shigella, with a reduction of lactic acid bacteria (e.g. Lactobacillus and Weissella) and short-chain fatty acid (SCFA) producers (e.g. Lachnospiraceae species) [17].
However, the impact of NE on the intestinal mycobiota of chickens is currently unknown. The purposes of this study were to investigate the ileal mycobiota changes in response to NE in broiler chickens and further reveal a possible correlation between the mycobiota and disease severity, laying a foundation for potential development of the mycobiome-based approaches to mitigating NE in poultry.
Materials and methods
Chickens and co-infection model of NE
Non-vaccinated day-of-hatch male Cobb broiler chicks were obtained from Cobb-Vantress (Siloam Springs, AR), tagged individually with wing bands, and assigned randomly to floor pens with fresh wood shavings. Chicks were provided ad libitum with tap water and an antibiotic-free corn-soybean starter diet (crude protein 21.5%) that meets or exceeds the nutrient requirements of the National Research Council (NRC) recommendations (1994). The lighting schedule was set as 23 L:1D in the first week and 18 L:6D afterwards. The room temperature was maintained at 32 °C in the first week and reduced to 30 °C and 27 °C in the second and third week, respectively. All animal procedures were approved by the Institutional Animal Care and Use Committee at Oklahoma State University under the protocol number AG-16-10.
On d 10, a total of 100 chickens were individually weighed after overnight fasting and transferred to 17 battery cages with 5–6 animals per cage for experimental induction of NE as previously described [18, 19]. Upon transfer, 90 chickens in 15 cages were immediately challenged with 5 × 103 sporulated oocysts of E. maxima strain M6 [20] in 1 mL saline via oral gavage, while the remaining 10 chickens in two cages were gavaged with 1 mL saline only and served as mock-infected controls. On d 14, after overnight fasting, 90 chickens that received E. maxima were orally inoculated again with approximately 4 × 108 colony-forming unit (CFU) of netBand tpeL-positive C. perfringens strain Brenda B [21] in 2 mL of overnight culture, which was prepared by sequential passage in cooked meat medium and fluid thioglycollate medium as described. Ten chickens in the mock-infected group were administrated with 2 mL fluid thioglycollate medium only.
All animals were monitored twice daily till d 17 for behavior and clinical signs. Mortalities were recorded daily and chickens reluctant to move were euthanized to alleviate undue pain. All surviving birds were weighed individually and euthanized through CO2 asphyxiation on d 17. Gross lesions of NE in the small intestine were evaluated in a blind manner using a 0–6 scoring system as proposed [18]. Briefly, the lesion scoring criteria were as follows: score 0 = no gross lesions, score 1 = thin or friable intestinal walls, score 2 = focal necrosis or ulceration (1–5 foci), score 3 = focal necrosis or ulceration (6–15 foci), score 4 = focal necrosis or ulceration (> 16 foci), score 5 = patches of 2- to 3-cm long necrosis, and score 6 = extensive necrosis typical of field cases. Contents from the proximal ileum (2–3 cm distal to Meckel’s diverticulum) were collected and stored at − 80 °C for microbial DNA extraction. Weight loss of infected chickens between d 10 and d 17 was calculated, relative to mock-infected controls.
Microbial DNA extraction
Microbial genomic DNA of the ileal contents was extracted using the ZR Fecal DNA MicroPrep Kit (Zymo Research, Irvine, CA) following the manufacturer’s protocol. The resulting DNA concentration and purity were quantified using Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) and used subsequently for microbial quantification and fungal ITS2 amplicon sequencing.
Quantification of total fungal and bacterial populations
Total populations of the fungi and bacteria in the ileal digesta were measured using Femto Fungal and Bacterial DNA Quantification Kits (ZYMO Research, Irvine, CA), respectively. Sample dilution and quantitative PCR (qPCR) were performed according to the manufacturer’s directions. Known quantities of purified genomic DNA of Saccharomyces cerevisiae and Escherichia coli were used to establish standard curves for fungi and bacteria, respectively. Total genome copies of the fungi or bacteria were estimated using the following formula as recommended by the manufacturer: genome copy number = DNA (g) / (g-to-bp constant × genome size), where the g-to-bp constant is 1.096 × 10− 21 g/bp and the average genome size of bacteria and fungi is 3.87 Mb [22] and 40 Mb [23], respectively. Results were expressed as fungal or bacterial genome copy number/g digesta, and the ratio of total genome copies of fungi to that of bacteria was further calculated for individual animals.
Fungal ITS2 sequencing and bioinformatics
Microbial DNA of the ileal contents was subjected to ITS2 sequencing for the mycobiota profiling. The ITS2 region was amplified by PCR using the primers ITS3-2024F (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4- 2409R (5′-TCCTCCGCTTATTGATATGC-3′) [24]. The ITS2 amplicon library was constructed using the NEBNext® Ultra™ DNA Library Prep Kit (New England Biolabs, Ipswich, MA) and subsequently sequenced on an Illumina HiSeq platform by Novogene (Beijing, China). PE250 paired-end reads were then processed using QIIME 2 v.2019.10 [25]. After demultiplexing and removal of adapters, sequence reads were denoised using Deblur [26] to generate amplicon sequence variants (ASVs). Taxonomic classification of fungal ASVs was implemented using Naive Bayes classifiers against the UNITE reference database (v.8.2). The taxonomies of top 30 and NEcorrelated fungal ASVs were confirmed by BLAST against the NCBI nucleotide database. The ASVs present in less than 5% of chickens were excluded from the analysis. The mycobiome sequencing data was normalized using the cumulative-sum scaling method in the R ‘metagenomeSeq’ package to correct uneven sampling depths [27].
Alpha and beta diversities of the fungal community were computed with R ‘phyloseq’ package (v.1.30.0) [28]. The number of ASVs, Pielou’s evenness index, and Shannon index were used to indicate the richness, evenness, and overall alpha diversity, respectively, whereas beta diversity was determined using weighted and unweighted UniFrac distances [29]. The mycobiota composition was indicated as relative abundances of fungal taxa at phylum, family, genus, and ASV levels. Fungal ASVs present in at least 20% of chickens were subjected to the linear discriminate analysis (LDA) effect size (LEfSe) analysis [30] to identify differentially enriched fungi between healthy and mildly-infected chickens as well as between mild and severe NE, with the cutoff at P < 0.05 and LDA score > 2.0. Spearman rank correlation analysis was performed between relative abundances of the fungal taxa existing in > 20% of chickens and NE severity indicated by lesion scores and weight loss. Spearman correlation coefficient was computed using the corr.test function in R ‘psych’ package (v.1.9.12.31) and displayed in Heatmap using the ‘pheat-map’ package (v.1.0.12) in R. Fold changes in relative abundance of significant NE-correlated taxa were calculated relative to that of mock-infected healthy chickens. Furthermore, ileal fungi-bacteria correlation was performed based on the Spearman correlation. NE severity-correlated fungal and bacterial taxa common in > 20% of chickens were included in the correlation analysis. The correlation matrix was plotted with R ‘corrplot’ package (v.0.84). Additionally, the ‘ggplot2’ package (v.3.3.0) [31] was used to make graphs in R.
Bacterial 16S rRNA gene sequencing and bioinformatics
Microbial DNA of the chicken ileal contents was also subjected to 16S rRNA gene sequencing for profiling the microbiota as previously described [32, 33]. The V4 region of the bacterial 16S rRNA gene was amplified by PCR using the primers 515F (GTGCCAGCMGCC GCGGTAA) and 806R (GGACTACHVGGGTWTCTA AT) and sequenced on Illumina MiSeq, processed with QIIME2, denoised with Deblur, and classified using the Greengenes database. The composition of the ileal microbiota was indicated by relative abundance of bacterial taxa at phylum, order, family, genus, and ASV levels. Those bacteria that were commonly present in > 20% of the samples were further calculated for their association with the lesion score using Spearman correlation analysis as described above for the mycobiome.
Statistical analysis
Statistical analysis and visualization were achieved in GraphPad Prism 8 (GraphPad Software, La Jolla, CA) and RStudio (v.1.2.1578) (RStudio, Boston, MA). Statistical significance was measured using parametric or nonparametric methods, depending on the normality of data as determined by the Shapiro-Wilk test. Weight loss, total fungi or bacteria, and the fungal/bacterial ratio were subjected to one-way analysis of variance (ANOVA) and Tukey’s post-hoc test, while alpha diversity and fungal relative abundance among groups were compared using Kruskal-Wallis and pairwise Wilcoxon rank-sum tests. The significance of beta diversity was measured by permutational multivariate analysis of variance (PERMANOVA) with 999 permutations using the R ‘vegan’ package (v.2.5.6). In Spearman correlation, the false discovery rate (FDR) was controlled using the Benjamini-Hochberg procedure. P < 0.05 or FDR < 0.05 was considered statistically significant.
Results
Growth impairment and ileal fungal load reduction by NE
As expected, sequential infections of chickens with E. maxima and C. perfringens induced clinical symptoms of NE including lethargy, anorexia, and diarrhea. Among 90 chickens infected, 33 died or were euthanized due to NE illness by d 17. All surviving chickens were scored for the severity of intestinal lesions using a 0–6 scale of a scoring scheme [18]. While the intestines of all 10 mock-infected chickens were apparently healthy and received a score of 0, all infected chickens had intestinal abnormalities, with lesions occurring primarily in the jejunum and proximal ileum. Among all infected chickens that were survived, 15 were scored 1, and 17 were given a score of 2. Five chickens were scored 5, while 13 birds were scored 6. Two infected chickens with a score of 3 and another two with a score of 4 were not included in the analyses because of the small sample size.
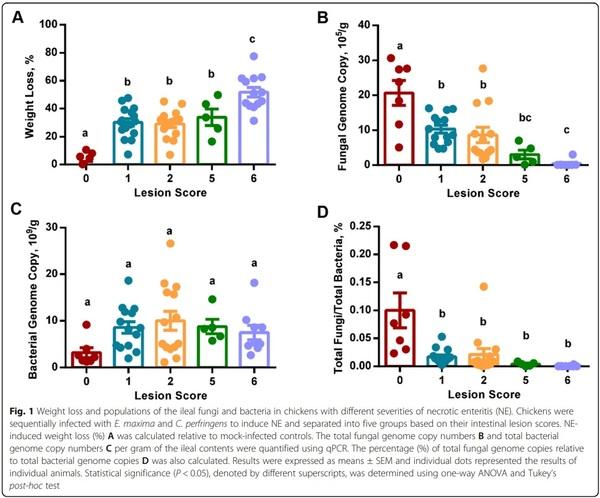
As a result, chickens were categorized into five groups (score-0, − 1, − 2, − 5, and − 6) based on their intestinal lesion scores. Although all groups of chickens had a similar body weight on d 10 prior to infection, weight loss of infected chickens was gradually increased as the lesions became more severe, with animals scored 1, 2, and 5 showing an approximately 30% weight loss as compared with mock-infected controls, while chickens scored 6 having a 52% reduction in weight gain between d 10–17 (Fig. 1A).
To evaluate the influence of NE on the intestinal microbial load, total fungal and bacterial populations in the ileal contents were quantified using qPCR. While mockinfected control (score-0) chickens had approximately 2 × 106 of fungal genome copies/g digesta, infected chickens showed a significant progressive decrease (P < 0.05) with exacerbation of NE (Fig. 1B). Chickens with a score of 1 or 2 harbored approximately 1 × 106 of fungal genome copies/g digesta; however, the fungal genomes were decreased to 3.0 × 105 /g digesta in chickens scored 5, which further declined to 4.3 × 104 /g digesta in severely infected chickens with a lesion score of 6 (Fig. 1B). By contrast, the genome copies of total bacteria were increased only by 2- to 3-fold in NE chickens, showing a plateau with chickens scored 2 (Fig. 1C). As a result, total fungal genome copies accounted for approximately 0.1% of total bacterial genome copies in the ileum of healthy broiler chickens, but was gradually declined in more severe NE (Fig. 1D). Total fungal population was decreased by approximately 5-fold to represent 0.02% of total bacteria in mildly infected chickens with a score of 1 or 2 and further reduced to only account for approximately 0.004% and 0.0006% of the bacterial population in more severe NE chickens with a lesion score of 5 and 6, respectively.
Alternations in the diversity of the ileal mycobiota in NE
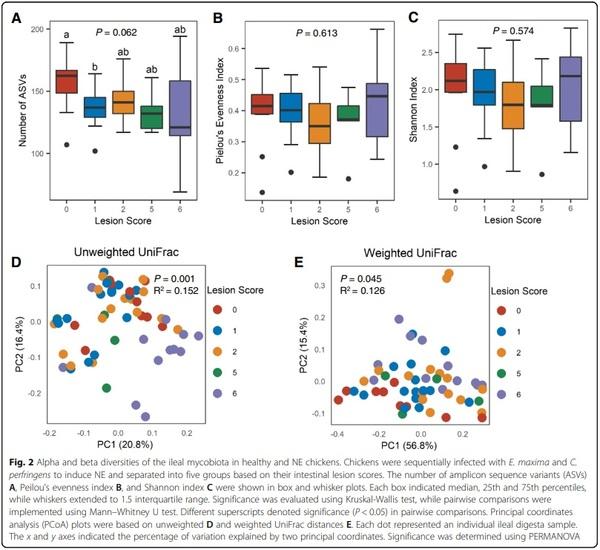
To investigate potential alternations of the intestinal mycobiome in NE, microbial DNA was isolated from the ileal digesta of both infected and mock-infected broilers and subjected to ITS2 sequencing. A total of 3,978,808 raw reads were obtained from 65 ileal digesta samples and analyzed using QIIME 2. After Deblur denoising, 1,512,578 sequence reads were left, with an average of 23,270 ± 1015 (SD) reads per sample, from which a total of 642 ASVs were generated. Samples with lesion scores of 3 and 4 were excluded from downstream analyses due to the small sample size. A total of 503 fungal ASVs present in > 5% of chickens with NE scores of 0, 1, 2, 5, and 6 were subjected to downstream analyses. Compared with healthy mock-infected (score-0) chickens harboring a median of 162 fungal ASVs, the fungal ASVs were gradually declined in NE chickens (P = 0.062) (Fig. 2A), indicating reduced richness of the fungal community in NE. Although Pielou’s evenness index was similar between healthy and all groups of NE chickens (Fig. 2B), the overall alpha diversity of ileal mycobiota measured by Shannon index was not changed obviously in NE chickens in comparison with healthy controls (Fig. 2C).
Comparisons of beta diversities of the ileal mycobiome between healthy and NE chickens revealed significant differences as indicated by unweighted UniFrac (P = 0.001, R2 = 0.152) (Fig. 2D) and weighted UniFrac (P = 0.045, R2 = 0.126) (Fig. 2E). Pairwise comparisons also revealed significant differences between score-6 chickens and other groups in unweighted UniFrac as well as significantly different weighted UniFrac distances between score-0 and score-6 chickens as well as between score-0 and score-2 chickens (P < 0.05) (Table S1). These results suggested that severe NE induced a pronounced shift of the ileal mycobiota.
Shifts in the mycobiota composition in the ileum of NE chickens
To further examine the ileal mycobiota compositional changes in response to NE, all fungi present in > 5% of the chickens were classified at phylum, family, genus, and ASV levels and compared among groups. The ileal mycobiota consisted of three phyla, 57 families, 90 genera, and 503 ASVs. The identities of top 30 fungal ASVs, averaging 92.3% of the total fungal population in all samples, were further confirmed by BLAST search of the NCBI nucleotide database (Table S2). Three phyla included Ascomycota, Basidiomycota, and Mucoromycota (Fig. 3A), with the predominant phylum Ascomycota comprising 73.5%–83.3% of the total fungal population, while the second most abundant phylum Basidiomycota constituting 15.8%–25.8% of the mycobiota (Table S3). However, none of the phyla showed a significant difference among different groups (Table S3). Wallemia was the only genus in the Wallemiaceae family and also the second most abundant genus in the ileum of chickens (Fig. 3B and C). Wallemia showed a gradual decline from 24.5% in healthy chickens to 10.1% in score-6 chickens (P = 0.02 and FDR = 0.08) (Table S3). By contrast, several other genera such as Pichia, Candida, Trechispora, Pseudotremella, and Malassezia were enriched in NE chickens in comparison with healthy controls (Fig. 3B, C, and Table S3).
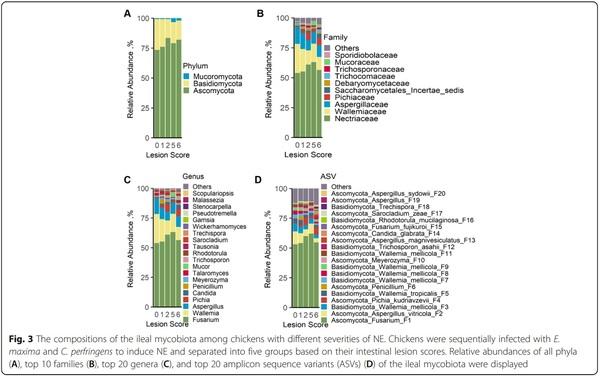
At the ASV level, a differential response of the ileal mycobiome to NE was observed. The most abundant ASV, an unidentified Fusarium species (F1) comprising 53.1%–62.2% of the total fungal population showed no obvious changes among healthy and infected chickens (Fig. 3D and Table S3). However, the second most abundant ASV, Aspergillus vitricola (F2), accounted for 10.7% of the mycobiota in healthy broilers and was gradually reduced to 3.5% in score-6 chickens (Fig. 3D and Table S3). Wallemia mellicola (F3, F7, F8, F9, and F11) and W. tropicalis (F5) also showed a progressive decline when NE was aggravated (Table S3). Conversely, Pichia kudriavzevii (F4) and Trichosporon asahii (F12) were more abundant in chickens with NE (Table S3).
Differential enrichment of the mycobiota in response to NE
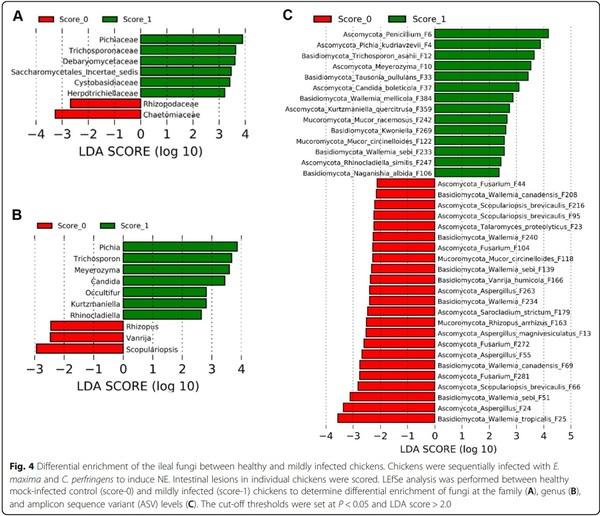
To identify discriminative fungi between healthy and NE chickens, LEfSe was first performed between score-0 and score-1 chickens at the family, genus, and ASV levels among those common taxa that were present in > 20% of chickens using thresholds of P < 0.05 and LDA score > 2. In comparison with healthy chickens, families such as Chaetomiaceae and Rhizopodaceae were reduced, while Pichiaceae, Trichosporonaceae, Debaryomycetaceae, Saccharomycetales incertae sedis, Cystobasidiaceae, and Herpotrichiellaceae were enriched in score-1 broilers (Fig. 4A). At the genus level, mild NE diminished Scopulariopsis, Vanrija, and Rhizopus, but enriched Pichia, Trichosporon, Meyerozyma, Candida, Occultifur, Kurtzmaniella, and Rhinocladiella (Fig. 4B). A total of 37 ASVs showed differential enrichment between healthy and mild NE chickens. Multiple members of Wallemia (F25, F51, F69, F139, F208, F234, and F240), Aspergillus (F13, F24, F55, and F263), and Fusarium (F44, F104, and F281), as well as Scopulariopsis brevicaulis (F95 and F216), were reduced in mild NE, while a diverse array of fungi such as a Penicillium species (F6). P. kudriavzevii (F4), a Meyerozyma species (F10), and T. asahii (F12) as well as two relatively rare Wallemia members (F233 and F384) were increased in mildly infected NE chickens (Fig. 4C). Fungi that were enriched in mild NE mainly belonged to Saccharomycetes, Tremellomycetes, and Chaetothyriales (data not shown).
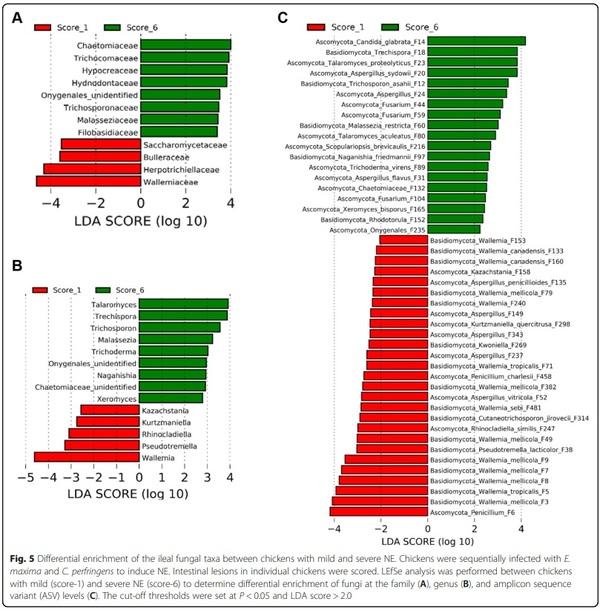

To further determine differentially abundant fungi in the ileum between chickens with mild and severe NE, LEfSe was performed between score-1 and score-6 chickens. At the family level, Wallemiaceae, Herpotrichiellaceae, Bulleraceae, and Saccharomycetaceae were enriched in chickens with mild NE, while Chaetomiaceae Trichocomaceae, Hypocreaceae, Hydnodontaceae, Trichosporonaceae, Malasseziaceae, Filobasidiaceae, and an unidentified family within Onygenales were more abundant in severely infected chickens (Fig. 5A). At the genus level, score-1 chickens showed an enrichment of Wallemia, Pseudotremella, Rhinocladiella, Kurtzmaniella, and Kazachstania, but genera such as Talaromyces, Trechispora, Trichosporon, Malassezia, Trichoderma, Naganishia, and Xeromyces were increased in score-6 chickens (Fig. 5B). At the ASV level, a total of 46 ASVs were differentially enriched between mild and severe NE. For example, Penicillium F6 and multiple Wallemia members such as W. mellicola (F3, F7, F8, F9, F49, F79, and F382), W. tropicalis (F5 and F71), W. canadensis (F133 and F160), and W. sebi (F481) were abundantly present in score-1 chickens, while enrichments of Candida glabrata, T. asahii, Aspergillus sydowii, and Aspergillus flavus were observed in score-6 chickens (Fig. 5C).
Correlation between the ileal fungal abundance and NE severity
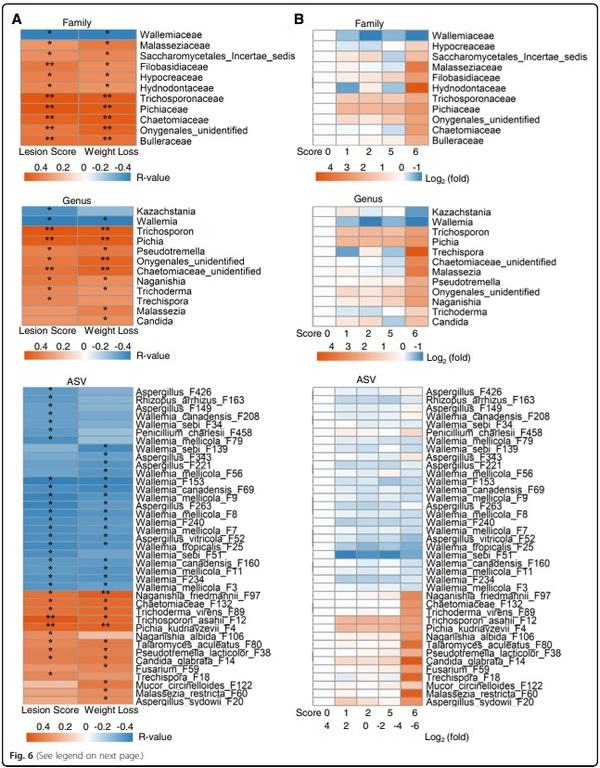
To identify the ileal fungi that are strongly correlated with NE severity, Spearman’s rank correlation was performed separately between the fungal taxa present in > 20% of the chickens and two NE severity parameters (intestinal lesion score and weight loss). Among 33 families and 47 genera that were common in > 20% of samples, 11 families and 12 genera showed a significant correlation (FDR < 0.05) with at least one indicator of NE severity (Fig. 6A). While Wallemiaceae/Wallemia was negatively correlated with disease severity (FDR < 0.05), Trichosporonaceae/Trichosporon, Pichiaceae/Pichia, Filobasidiaceae/Naganishia, an unidentified member of Onygenales, and Bulleraceae/Pseudotremella showed a significant positive correlation (FDR < 0.05) with both NE severity indicators. Out of 198 fungal ASVs present in > 20% of chickens, 39 ASVs showed a significant correlation with NE severity, with 14 showing a positive correlation and 25 showing a negative correlation (FDR < 0.05) (Fig. 6A). For example, 17 Wallemia species such as W. mellicola (F3, F7, F8, F9, and F11), W. tropicalis (F25), W. sebi (F34 and F51) and six Aspergillus members (e.g., A. vitricola F52 and unclassified Aspergillus) exhibited a strong negative correlation with at least one indicator of NE severity (Fig. 6A). As revealed in a heatmap, multiple Wallemia members were diminished, while a diverse group of fungi were enriched in exacerbated NE (Fig. 6B).
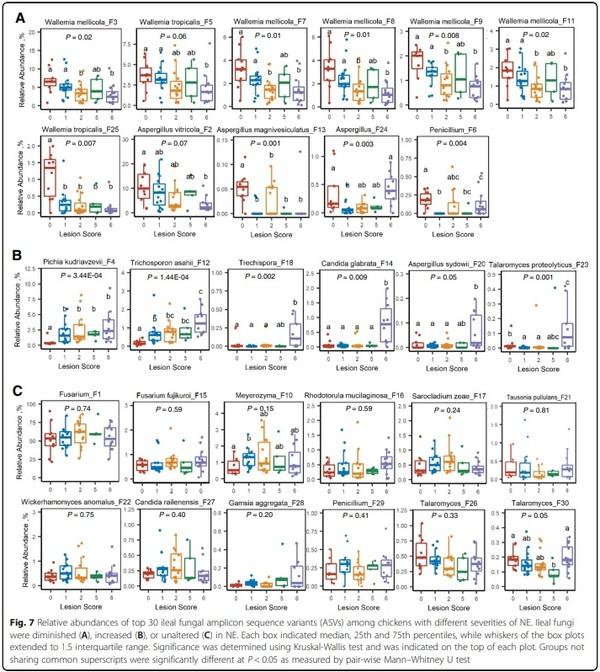
A closer examination of the 30 most abundant fungal ASVs further revealed different patterns of the fungal change in response to NE. While W. mellicola (F3, F7, F8, F9, and F11) and W. tropicalis (F5) were gradually diminished in more severe NE; W. tropicalis F25 showed an abrupt decrease even in chickens with mild NE (Fig. 7A), suggesting both inter-species and intra-species variations of Wallemia in response to NE. Similarly, different sensitivities to NE were observed among Aspergillus species. While A. vitricola (F2) displayed a progressive decline, A. magnivesiculatus (F13) was abruptly abolished in all NE chickens (Fig. 7A). On the other hand, A. sydowii (F20) appeared to be enriched only in chickens with severe NE (Fig. 7B). An unspecified Trechispora (F18), C. glabrata (F14), and Talaromyces proteolyticus (F23) were also sharply increased in severe NE chickens (P < 0.05), but P. kudriavzevii (F4) and T. asahii (F12) showed a progressive increase with NE severity (Fig. 7B). Notably, nearly 50% of top 30 fungal ASVs showed no obvious shift change in response to NE including the most abundant Fusarium ASV (F1) (Fig. 7C). Perhaps as confirmation, another Fusarium (F. fujikuroi F15) was unaltered in NE either (Fig. 7C).
Correlation between the ileal mycobiota and microbiota in the context of NE
In addition to the mycobiota, the bacterial microbiota of the ileum was also investigated by 16S rRNA gene sequencing. Spearman correlation analysis was conducted between the ileal fungi and bacteria to explore their potential interactions in the context of NE. Among 12 fungal and 29 bacterial genera that were commonly present in > 20% of the chickens and also showed a significant correlation with NE severity (FDR < 0.05), Wallemia showed a negative correlation with Clostridium, but a positive correlation with Cyanobacteria, Subdoligranulum, Corynebacterium, Blautia, Staphylococcus, Cuneatibacter, Aerococcus, Lactonifactor, Oscillospiraceae, Erysipelatoclostridium, and an unidentified genus in Lachnospiraceae (Fig. 8A). Positive correlations also occurred between Clostridium and fungal genera including Pichia, Candida, Trichosporon, Pseudotremella, and Naganishia, while a majority of the fungal-bacterial interactions were negative (Fig. 8A).
At the ASV level, among 39 fungal and 64 bacterial ASVs showing a significant correlation with the severity of NE (FDR < 0.05), positive correlations were common among NE-diminished fungi such as Wallemia species and NE-reduced bacteria (majorly lactic acid bacteria and SCFA-producing bacteria, such as group B Lactobacillus, L. reuteri, Subdoligranulum variabile, Weissella species, and Blautia species) (Fig. 8B). On the contrary, negative interactions mainly occurred between NE-enriched fungi (e.g., P. kudriavzevii F4, T. asahii F12, C. glabrata F14, and Naganishia friedmannii F97) and bacterial species that were decreased in NE (Fig. 8B).
Discussion
The bacterial microbiota and fungal mycobiota residing in the GI tract contribute to the health or diseases of the host [1–5]. Disturbance in the intestinal microbiota has been linked to chicken NE [17], while the involvement of the mycobiome in NE is yet to be investigated. In the current study, we unraveled an altered ileal fungal community in NE-afflicted chickens using ITS2 amplicon sequencing and further identified a number of fungi that are strongly correlated with the severity of NE. Our study also revealed the mycobiota-microbiota correlations in NE, suggesting that the mycobiome, in addition to the microbiome, might be potentially involved in NE and targeted to mitigate NE, although direct experimental evidence is needed.
We found that chicken intestinal mycobiota is dominated by Ascomycota followed by Basidiomycota. The two fungal phyla make up 97%–99% of the ileal mycobiota in broilers, in agreement with a recent report [14]. Similarly, Ascomycota and Basidiomycota are two major fungal phyla found in the human GI tract [8, 9]. Ascomycota is also the most abundant fungal phylum in the intestine of piglets [34]. In the present study, Fusarium is the most predominant in the chicken ileum, regardless of the health status. Fusarium, Wallemia, and Aspergillus collectively comprise 84% of the ileal fungal community, while Pichia, Candida, Penicillium, Mucor, and Trichosporon also show a high prevalence in the chicken ileum. Similar to our results, Candida, Trichosporon, and Rhodotorula are major fungi isolated from chicken feces [35], which are also among predominant fungi in the GI tract of healthy turkeys [36]. As for the source of the intestinal fungi, Fusarium, Aspergillus, Penicillium, and Mucor were found to be prevalent in corn, soybean meal, and finished poultry feed [37, 38], suggesting that the intestinal mycobiota is mainly originated from the feed.
In this study, we found that richness, but not evenness or Shannon index, of the ileal mycobiota tends to decrease in NE, which is similar to earlier studies with IBD and ulcerative colitis patients [3, 8], although other studies described no obvious changes in fungal richness between Crohn’s disease patients and healthy cohorts [13, 39]. One major finding of our research is that the total ileal fungal population is drastically reduced in severe NE. The fungal population approximately constitutes approximately 0.1% of the total bacteria in the ileum of healthy chickens, which is consistent with the previous report in humans and mice [40], but is gradually diminished in NE, with an approximately 50-fold reduction in severely infected, score-6 chickens. On the other hand, the total bacterial load is increased by 2- to 3-fold in NE chickens. As a result, a progressive decline in the fungal-bacterial ratio occurs in exacerbated NE. Interestingly, the fungal load and the fungal-bacterial ratio are increased in patients with Crohn’s disease [39]. More research is warranted to further investigate whether colonization and proliferation of C. perfringens in the small intestine in NE lead to diminished mycobiota.
In this study, we observed an obvious difference in the sensitivity to NE among the ileal fungal taxa. Among the 30 most abundant fungi, approximately 40% remain unchanged, while another 40% are declined and the remaining 20% are enriched in NE. Among those that are altered in NE, some show a gradual increase or decrease, while others are changed abruptly. For example, C. glabrata (F14), A. sydowii (F20), T. proteolyticus (F23) and a Trechispora species (F18) are enriched only in the ileum of severally infected chickens.
Among those fungi that are dramatically reduced in NE are a number of Wallemia taxa that are commonly found in the air, house dust, soils, and plants [11, 41]. Wallemia is also a commensal in the intestine of humans and mice [11]. In this study, we revealed that several Wallemia species such as W. mellicola, W. tropicalis, W. sebi, and W. canadensis are negatively correlated with NE severity. Consistently, Wallemia is reported to produce UCA1064-A and 1064-B with beneficial antitumor, antifungal, and antimicrobial activities [42, 43]. However, W. sebi and W. mellicola have been found to be associated with skin infections and allergic airway diseases [10, 11]. Further studies are needed to understand the involvement of Wallemia in NE.
Apart from Wallemia, Aspergillus is also apparently reduced in NE. A. vitricola, A. magnivesiculatus, and several unidentified Aspergillus species are negatively correlated with NE severity. Aspergillus is ubiquitous in feed and can cause aspergillosis in avian species or humans [44]. Although it can be a source of dietary mycotoxins with a detrimental effect on poultry health and performance [37], Aspergillus also produces beneficial metabolites such as lovastatin, terreulactones, 11-α hydroprogesterone, quadrone, and terpeptin [45]. The potential of Aspergillus for disease resistance in poultry warrants further investigation.
Along with a striking decrease in Wallemia and Aspergillus, other fungi such as P. kudriavzevii (teleomorph of Candida krusei), C. glabrata, M. restricta, T. asahii, and N. friedmannii are enriched in the ileum of chickens with NE, especially in severe NE. P. kudriavzevii isolated from chicken feces exhibits probiotic properties in vitro [46]. With a capacity to bind to aflatoxin B1, dietary supplementation of P. kudriavzevii mitigates the adverse effect of aflatoxin B1 on the growth performance of broilers [47]. Candida species are a part of commensal mycobiota in the GI tract but may cause candidiasis in poultry and humans [36, 41, 44]. Candida infections most frequently occur in the upper GI tract of chickens and may result in growth retardation or even mortality [44]. We revealed a positive correlation between C. glabrata and NE severity, which is in agreement with reports that Candida such as C. albicans, C. glabrata, and C. tropicalis are associated with IBD [8, 13, 39]. The role of P. kudriavzevii and Candida overgrowth in NE is not clear, and further studies are warranted.
Malassezia is a commensal fungus that colonizes not only the skin but also in the GI tract of humans and animals [48]. M. restricta has been found to be associated with IBD and several other intestinal inflammatory disorders in humans [48]. M. restricta, with potent pro-inflammatory properties, is enriched in Crohn’s disease patients, and oral administration of M. restricta exacerbates colitis in mice [9]. T. asahii is another commensal fungus that can cause opportunistic infections [14, 49]. In agreement with expanded intestinal Trichosporon in IBD patients [8], we found that T. asahii is positively correlated with NE severity in chickens. In addition, N. friedmannii (formerly Cryptococcus friedmannii) is reported to cause onychomycosis in humans [50]. The overgrowth of opportunistic fungi in NE-infected chickens might increase susceptibility of chickens to mycosis and probably threat public health.
Fungi and bacteria co-colonize the GI tract and interact with each other directly or indirectly through physical contact, microbial metabolites, and modification of immune status [4, 51]. The cross-talk between the mycobiota and the microbiota is critical for maintaining intestinal homeostasis [4, 51] and has been demonstrated in turkeys [15] and human IBD [8, 13]. We revealed a dramatic shift of the ileal microbiota in NE (unpublished). The current study has further revealed a strong positive or negative correlation between a number of fungal and bacterial taxa in NE. For example, most Wallemia species are correlated negatively with C. perfringens colonization in the ileum but correlated positively with a number of SCFA-producing bacteria, consistent with an earlier report on a positive association between Wallemia and SCFA-producing Oscillospiraceae [52]. On the other hand, P. kudriavzevii (F4), T. asahii (F12), C. glabrata (F14), and M. restricta (F60) are positively correlated with C. perfringens with a negative correlation with SCFA-producing bacteria and often lactic acid bacteria. Such an antagonism between C. glabrata and lactic acid bacteria (e.g., Lactobacillus and Weissella) was also reported earlier [53, 54]. It will be important to study the role of the fungi-bacteria interplay in the development of NE and whether such interactions can be explored for control and prevention of NE.
Eimeria infection is an important predisposing factor for NE in poultry with the ability to cause damage to the intestinal epithelium, providing niches or nutrients to facilitate the colonization and proliferation of C. perfringens [18]. Eimeria in conjunction with C. perfringens challenge is thus the most commonly used approach to experimentally induce NE, while the same dose of Eimeria or C. perfringens alone causes no or only mild intestinal lesions [18]. Consistently, co-infection with Eimeria and C. perfringens causes more pronounced microbiota changes than inoculation separately with Eimeria or C. perfringens [55, 56]. However, the impact of Eimeria or C. perfringens on the intestinal mycobiota is currently unknown and warrants further investigation.
Conclusion
This study revealed for the first time dysbiosis of the chicken ileal mycobiota induced by NE. The total fungal population is drastically reduced in NE and alterations in the mycobiota are more pronounced in exacerbated NE. Furthermore, we reported positive and negative correlations between a number of fungi and bacteria. These findings suggest a possible role of the intestinal mycobiota in NE pathogenesis and highlight the mycobiota as a new potential target for NE management in poultry.