1. Introduction
Awareness of mycotoxins has grown mainly in the last fifty years since the discovery of aflatoxins in the 1960s, although they have accompanied mankind from the very beginnings and were probably associated with several mysterious diseases known from history [1]. Mycotoxins are now recognized as prevalently toxic compounds produced as secondary metabolites by various fungi and excreted into their substrates. These substrates frequently include plants grown and stored for human or animal consumption as well as processed food. Mycotoxins and the associated health disorders in humans and animals have been recognized as a major health and economical problem [1, 2], which dictates measures to minimize the exposure by applying proper agricultural practice, storage of products and control of the products intended for human or animal consumption [3–10]. Moreover, there is also a growing awareness of the mycotoxin presence in the living and working environment and associated disease [11-14]. The control measures to ensure mycotoxin-free food, feed and environment imply chemical analysis of these contaminants in a great variety of samples, further complicated by the structural diversity of mycotoxins which call for different analytical methods.
The aim of the present review is to present the state-of-the-art of the conventional and emerging analytical and sample preparation methods for the most important mycotoxins in various matrices where they may be encountered. Besides, screening methods are also covered. Special emphasis is on the pitfalls of the mycotoxin analysis and the errors that may be inherent or introduced in each step of the analytical method. As this is a very broad scope, the review covers only the methods and techniques published in the last decade.
1.1. Diversity and impact of mycotoxins
Various commonly found types of molds produce and excrete mycotoxins. Between 300 and 400 different mycotoxins are known today, but not all of them are present in higher concentrations or have a significant health or economical impact and therefore have to be determined in various matrices [15, 16].
Table 1 lists the major groups of mycotoxins or individual compounds that are the most interesting from the analytical point of view as stated above. The abbrevations in the Table 1 are used throughout the text. Moreover, fungal species that produce them and the most common health disorders they cause are also listed. The chemical structures of these compounds are shown in Figures 1–6. There are other toxicologically important mycotoxins not included in Table 1 as they are less frequently encountered, e.g. penitrems, thomitrems, lolitrem, verruculogen, griseofulvin, chaetoglobosin, sambutoxin, citreoviridin, apicidin, roridin, monacolin K, phomopsins, sporidesmins, AAL toxins, satratoxins etc. [1, 2, 12, 17].
The health disorders listed in Table 1 are those most frequently associated with the exposure, but especially the data on acute toxicity of some mycotoxins show considerable variation in the available studies [1, 2, 7, 16, 18]. The susceptibility of animals (and humans) varies with species, age, nutrition, length of exposure and other factors as well [1, 2]. Evaluation of adverse health effects is complicated by co-existence of various mycotoxins in food and feed and their possible synergistic action [16, 19]. Many mycotoxins exhibit a rather non-specific action at the usual exposure levels, e.g. immunosuppression [2] and consequently can increase susceptibility to other illnesses [7]. Although it has been postulated that several unexplained disease outbreaks from the past were caused by the consumption of mycotoxin-contaminated food [1, 2, 7, 20], e.g. Balkan endemic nephropathy [19, 21], other environmental factors such as drinking water contamination may also play a role in their occurrence [22]. At present, there is no firm evidence that the normal daily exposure to some mycotoxins listed in Table 1 (RQ, MPA) would lead to health disorders [23, 24] or these occur only at higher intake levels (e.g. for PAT) [25], but nevertheless the maximum tolerable daily intake and residual levels in foods have been established [1, 25].
Figure 1. Structures of aflatoxins {AFs} B1, B2, M1, G1, G2.
Figure 2. Structures of some important trichothecenes.
Figure 3. Structures of enniatins.
Figure 4. Structures of some Alternaria mycotoxins: alternariol {AOH} (1), altenuene {ALT}(2), altertoxin I{ATX-I}(3).
Figure 5: Structures of some Fusarium mycotoxins: FUM B1 (1), moniliformin (usually as sodium or potassium salt) (2), zearalenone {ZON} (R =O) and zearalenol {ZOL} (R -OH) (3), possible metabolite zearalanone {ZAN} (4).
Figure 6. Structures of some important mycotoxins produced by Penicillium and Aspergillus species: ochratoxin A {OTA}(1), patulin {PAT}(2), citrinin {CIT} (3), cyclopiazonic acid {CPA} (4), roquefortin C {RQ} (5), mycophenolic acid {MPA}(6).
1.2. Diversity of samples containing mycotoxins
Molds and their metabolites are present everywhere in the environment, which implies a wide variety of samples that might contain mycotoxins and have to be analyzed. Table 2 lists the samples an analytical chemist will most frequently encounter. The detailed analytical approaches performed for these samples, however, are discussed in Section 5 of this review.
Because of the deleterious health effects of mycotoxins, their levels have been strictly regulated especially in food and feed samples. While European Union has in general set somewhat lower allowable residue levels than US Food and Drug Administration, they are usually in the μg/kg–mg/kg range for the majority of compounds [1, 4]. Various types of grains are the most burdened in terms of different possible compounds, mostly occurring pre-harvest and some also post-harvest during storage, although the latter is easier to control [1, 2, 4, 20, 26]. The first evaluation of food and feed is by inspection for the visible fungal contamination as well as for the characteristic moldy odor, although these factors don’t necessarily correlate with the mycotoxin concentration and are also not applicable to processed foods.
In samples of animal origin, such as meat, milk, dairy products and eggs, mostly aflatoxin metabolite M1 is expected and also OTA in some tissues (liver, kidney). Other mycotoxins haven’t been shown to exhibit carry-over from feed to animal products [1, 3]. Cheese deliberately inoculated with presumably non-toxicogenic molds may in addition contain MPA, RQ [23, 24] and to a certain extent also CIT and CPA are expected [19, 27]. Fish may contain RALs [28]. Special care has to be taken while evaluating mycotoxin content in processed foods, as during processing, especially heating, the content of mycotoxins is generally reduced, but degradation products of both higher or lower toxicity may form [9, 19].
Another group of samples in Table 2 are from the indoor environment. They have been gaining in importance since the recognition of the “sick building syndrome”, a chronic illness associated with fatigue, allergic reactions, neurological symptoms and higher incidence of cancer. Affected people live in mold-infected buildings with fungal spores containing mycotoxins present in the air and house dust [11, 12], but can be exposed also in the working environment [13, 14]. Analysis of human samples (blood, urine, milk) is therefore also important to evaluate the extent of exposure from environment and food [29, 30].
Finally, environmental samples such as soil and environmental waters may provide information on the fate of mycotoxins formed in fungally infected agricultural crops [31, 32] or of anthropogenic origin [33].
2. Screening methods
Different types of samples and their vast number, as well as chemical diversity of mycotoxins and their simultaneous occurrence in samples present a need for rapid multi-analyte methods suited for various matrices. Moreover, they should be sensitive enough to detect mycotoxins below the legally imposed limits. Separation methods coupled to sensitive and/or selective detection methods fulfill these requirements and are at this time the most definitive, but their drawback is that they are preceded by time-consuming sample extraction and clean-up, except if the raw extracts are analyzed (e.g. with LC-MS/MS). They are also rather expensive and demand specially trained personnel. For these reasons, the traditional as well as novel screening methods have found a wide application area in mycotoxin analysis [34]. They usually provide rapid and sensitive detection, are very cost-effective and easy to use, thus they can be used by non-specialists and under field conditions as well. Although there is a greater emphasis on selectivity in novel assays [34], the main drawback of many screening methods is still their cross-reactivity. Positive results should therefore be confirmed with more selective methods to avoid misinterpretations [35].
2.1. Immunoassay-based methods
These methods are based on the interactions between antibodies and antigens, which are in this case mycotoxins. Antibodies are highly specific for structurally very different compounds, but can show considerable cross-reactivity for structural analogs, as they recognize only specific chemical groups - the epitope [15, 36]. Mycotoxins are small, non-immunogenic molecules (haptens) and have to be bound to suitable carrier proteins to elicit adequate immune response and production of antibodies in an animal [37]. Novel methods of recombinant antibody production can circumvent the binding of hapten to protein and inherent risk that the free hapten would not be detected in samples, as well as animal immunization [38]. Another compound, variously named marker, tracer or label, is involved in the assay to facilitate detection. The marker may be radioactive in radioimmunoassay (RIA) - rarely used now, or a chromogenic or fluorogenic compound reacting with enzyme in enzyme immunoassay (EIA, ELISA) or in fluorescence immunoassay (FIA), respectively.
Table 1. Major mycotoxin groups or individual compounds, the fungal species producing them and health effects they cause. Symbols for mycotoxins are written in {} curly brackets. NUA denotes “not unequivocally established”.
ELISA (enzyme-linked immunosorbent assay) is the most frequently applied type of assay [39, 40]. Substrate for the enzyme is usually a chromogenic substance. The assay is mostly performed in a 96- well plate, allowing simultaneous analysis of up to 45 samples in duplicate. Incubation times are 0.5–2 h and the developed color is usually measured spectrophotometrically [41]. Other types of detection are possible, e.g. amperometric [42] or by differential pulse voltammetry [43]. The most simple, however, is the visual comparison of color intensity, providing either semiquantitative results [44] or a yes/no response at a certain concentration level [34] or concentration range [41].
Other immunoassay type employs fluorescent markers, which involve fluorogenic substrate reacting with enzyme-linked analyte [34, 40, 45, 46] or measure fluorescence polarization induced by increased molecular mass of antibody-bound labeled mycotoxin [34, 39, 40, 41, 47].
Marker-free immunoassays are based on natural fluorescence of some mycotoxins (AFs, OTs), conductometric or impedimetric measurements [34]. Surface-plasmon resonance-based immunoassays (SPR) employ the change of surface optical characteristics upon binding of antibodies to the surfacebound antigen. The amount of free antibodies correlates with the mycotoxin content of the sample with which they are incubated [34, 39, 41, 48, 49].
Besides the various detection modes, different constructions have also been developed in the field of immunoassay methods. The most common design is a multi-well microtiter plate used in classic ELISA with spectrophotometric or electrochemical detection [50]. Flow-through or flow-injection immunoassays use immobilized proteins to separate antigen-antibody complexes and detect only the free marker [40, 42, 45]. Mycotoxin-tagged liposomes have been prepared to develop a flow-injection liposome immunoanalysis (FILIA) [51]. Flow-through test may be used as another name for immunofiltration, where antibodies are immobilized on a semi-permeable membrane. Such devices are simple to use in field conditions with visual detection, but can be also quantitatively evaluated with instrumental detection in laboratory settings [41, 44]. Finally, various immunostrips or immunodipsticks, also named lateral flow devices (tests), have been developed for mycotoxin testing in the field, using either membrane with immobilized antibodies or dry reagents on the test strip. In the latter case, the sample would reach and dissolve the reagents by lateral capillary flow and the test is thus self-developing [34, 39-41].
With respect to mycotoxin types, various innovative immunoassays mentioned above were developed for most of the main groups: AFs [34, 42-44, 47, 50], trichothecenes [34, 41], FUMs [34, 38, 48, 51], RALs [34, 45], OTA [34, 37] and PAT [46]. On the other hand, immunosensor arrays, designed to detect several mycotoxins from different groups simultaneously, are gaining in importance as well [34, 41, 49, 52-54].
Immunoassays with colorimetric and fluorimetric detection generally display favorable detection limits (LOD 1-20 μg/kg) [34, 40, 55], but it must be re-emphasized that they should be used as a screening tool only to detect possible contamination with mycotoxins. It has been established that while false-negatives are rare, false-positives are more frequent and depend on many factors. Besides the inherent cross-reactivity, other conditions influence the result: temperature, sample viscosity, pH, ionic strength [41]. If no sample clean-up or extraction is performed before the test, matrix effects might be expected [39, 55], sometimes leading to significant overestimation of mycotoxin concentration [39, 56]. Extraction usually involves polar organic solvents, although they are suboptimal for most mycotoxins. Typically methanol is used, which is tolerated to a various percentage depending on the design of the immunoassay [54]. Co-extracted substances, e.g. fat, may also cause problems. For water-soluble mycotoxins, water or buffers can be used instead [41], but in any case, additional clean-up may be needed, especially for colored samples [57]. In case of visual estimation of tests, a concentration range should be considered rather than a cut-off value which is difficult to establish due to the analog nature of both color development and human vision [41]. For all the reasons listed above, samples shown to be positive on mycotoxins should be re-analyzed by the reference method.
2.2. Sensors and biosensors
Sensors are devices composed of two elements: the molecule (usually biological), which selectively reacts with the compound(s) of interest (recognition element) and is in contact with a transducing element converting the change of the physical variable produced by the reaction into a measurable signal. In this respect, biosensors are those using biological molecules/entities as recognition elements: antibodies, enzymes, bacteria, receptors, DNA [34, 58, 59]. Transducing elements are usually optical or electrochemical.
Some of the immunoassay approaches described in the previous section can be considered as biosensors, provided that the measured signal originates directly from the antibody-antigen complex in contact with the transducing element [34]. The latter can be an optical detector based on surface plasmon resonance [40, 48, 49, 60], fluorescence [61], optical waveguide lightmode spectroscopy - OWLS [53] or total internal reflection ellipsometry - TIRE [34]. Another possibility is an electrochemical detector based on potentiometry with carbon working electrode [50, 62], differential pulse voltammetry, conductometry [34], chronoamperometry [63] and electrochemical impedance spectroscopy [64]. Some of these sensors are for single mycotoxins and others are made for multitoxin sensing (bisensor array). Other types of biosensors include enzymatically-mediated electrochemical sensors based on potentiometry, and DNA sensors [34, 59].
The advantages and disadvantages of biosensors are essentially the same as those of immunoassays: they are cheap, fast, portable, very suitable for routine screening of samples, but they suffer from the selectivity and reproducibility problems. The results should be therefore confirmed with the reference method [34, 58]. However, some electrochemical biosensors show excellent sensitivity with LODs below 0.1 μg/kg [34, 50, 63], while those based on optical detection achieve LODs comparable with ELISA (0.5-10 μg/kg) [34, 53].
Another type of sensors that mimic the biochemical recognition molecules is the one based on molecularly imprinted polymers (MIPs) [34, 40, 65-68]. These functional materials have found a wider application in the field of selective sample preparation and will be discussed in more detail in the next section. MIP-based sensors have some advantages over biosensors: better stability in organic solvents, decreased degradation and loss of active sites [34, 65], but so far only the sensors with fluorescent tracers displaced by the analyte have been designed [67]. Problems of these sensors are connected with problems of MIP materials in general: selectivity [34, 67], reproducible preparation [68], sample contamination [66, 68]. Nevertheless, MIP-based sensors are very promising and are expected to find wider application in the future.
2.3. Other direct screening methods
Several approaches making use of the different chemical and physical characteristics of mycotoxins will be mentioned here: some already established, others just emerging. The best known method of the former is thin-layer chromatography (TLC), which, although used also for the more accurate analyses, can be applied for the screening purposes as well with visual or densitometric detection. Compared to ELISA, it shows better repeatability and is less prone to overestimation of the concentration due to its better selectivity, but requires more extensive sample clean-up and is more time-consuming [69].
Flow-injection systems use a flow-through detector with rather high specificity for the analytes of interest. Examples are the system for total AFs using fluorimetric screening [70] and system for RALs using electrochemical detection [71]. In both cases an extraction and clean-up step is required. In case of samples with total concentration close to or above threshold limit, confirmation analysis is needed with a more selective method [70, 71]. Static fluorimetric analysis of previously extracted and derivatized analyte was applied for DON screening in grains [72].
An interesting AFB detection method is based on acetylcholinesterase inhibition by AFB1, subsequent enzyme substrate addition and spectrophotometric measurement of the product. The assay is very fast and quite sensitive, but strongly depends on the temperature and methanol concentration. It also shows a significant cross-reactivity to other AFs [73]. Another bioassay for trichothecenes was developed, exploiting the inhibition of enzyme β-galactosidase in the yeast with subsequent colorimetric detection of the product of enzymatic reaction. With the additional use of the toxicityenhancing agents, LODs comparable with ELISA were achieved [74].
2.4. Indirect screening methods
These methods are based on the measurement/determination and interpretation of a parameter or compound(s) in the sample that change relative to the mycotoxin concentration. The relation has to be established on a series of samples analyzed by a reference method. Most of these methods are also non-invasive, meaning less of the sample manipulation and faster throughput.
Various methods using infrared spectroscopy have been developed, e.g. Fourier transform midinfrared spectroscopy (FTIR) in diffuse reflection (DR) [75] or attenuated total reflection (ATR) mode for determination of DON in wheat or maize [39, 75, 76]. Principal component analysis (PCA) and cluster analysis were applied to evaluate the spectra and classify them on the basis of the mycotoxin contamination [75-77]. Spectra obtained in ATR mode were superior to DR spectra in terms of classification efficiency, e.g. samples with DON concentration 2.5-12.1 mg/kg were in 100 % classified as contaminated [75]. FTIR-ATR measurements with partial least squares (PLS) as a regression method were successfully applied to evaluate OTA contamination in dried grapes [78, 79]. Near-infrared spectroscopy (NIR) in the transmission mode with PCA and PLS calculations were used to develop a screening method for DON in wheat [39, 80]. NIR in reflectance mode and modified PLS algorithm were applied to determine total AFs, AFB1 and OTA in red paprika [81]. All of the above methods are applied without any sample preparation except milling and sieving [77], meaning great savings in time, but the disadvantage is that they are valid only for the sample matrix used during calibration [39]. Even if this condition is met, poor spectral reproducibility may still occur for samples of the same type, but from different regions [78].
A quite different approach for indirect detection of mycotoxins in sample is detection of volatile metabolites of the fungi. In this respect, a differentiation should be possible between toxicogenic (mycotoxin-producing) and non-toxicogenic strains. Determination of volatiles produced by fungi may
be performed with the established methods, e.g. GC-MS with previous headspace extraction [82, 83], but there is an increasing number of publications where the development of an electronic nose (e-nose) for this purpose is described. E-nose is an array of gas sensors based mostly on metal oxides. Since the response is non-specific, it has to be calibrated against reference method for volatiles and processed by chemometric methods to be able to classify subsequent samples in the appropriate groups [84-88]. So far, this approach has been successfully used for the determination and even prediction of concentration of OTA (classification limit 5 μg/kg) and DON in barley [87] and PAT in apples [86]. Other publications report only detection of mold growth on wheat [84, 85], the approach that can be as effective as sensory panels [88]. However, it is possible to differentiate between toxicogenic and nontoxicogenic strains of fungi by detecting specific volatile metabolite(s), albeit as far as now only by GC-MS [82, 83]. Again, sample preparation step is shortened or even absent in the case of e-nose use, but matrix dependence is significant in this approach as well.
As an estimation of the total fungal biomass in samples, e.g. maize, the determination of fungal metabolite ergosterol is often performed with HPLC [76, 89, 90].
3. Sample preparation and pre-concentration
One of the crucial steps for the qualitative and quantitative determination of individual mycotoxins is the sample preparation and pre-concentration. Several different extraction procedures are used for the purpose and will be overviewed in this section. Some of these are well-established techniques and others just emerging, which have yet to prove their relative advantages and demonstrate shortcomings. The emphasis in the field of sample preparation in the last few decades has been on the minimization of solvent use, especially of environmentally and health-harmful chlorinated solvents, as well as on the miniaturization of the procedures, meaning also smaller sample size, while at the same time maintaining the efficiency. The other field of development is in the highly analyte-specific extraction techniques with added benefit of reduced need for subsequent purification of the extracts. On the other hand, older sample preparation techniques, although usually time-consuming and more labor-intensive, have the advantage of long-term use: the possible pitfalls are well recognized and can thus be relatively easily avoided. One should bear in mind that several serious systematic errors are possible in this step of analysis, some of which will be mentioned shortly, and then again overviewed in the Section 6 of this review.
3.1. Solvent extraction
The oldest, but still frequently used sample preparation technique is solvent extraction (SE), in the case of liquid samples also named liquid-liquid extraction (LLE). Both aqueous - e.g. buffers, hot water, and organic solvents - e.g. acetonitrile, chloroform, methanol, ethyl acetate etc. are used. Samples may be solid or liquid. With liquid samples this step serves as an enrichment and/or clean-up procedure, while with solid samples SE is a necessary pre-step for further manipulation of sample extract. Different modes of SE can be employed, such as conventional Soxhlet extraction, used for solid samples [91] and ultrasonic extraction [92]. One well-recognized drawback of Soxhlet extraction is the possible production of artifacts from thermally less stable compounds, although it hasn’t yet been shown for mycotoxins. More recent techniques include supercritical fluid extraction (SFE) [71], pressurized liquid extraction (PLE), also known as accelerated solvent extraction (ASE) [45, 93-96] or microwave-assisted extraction (MAE) [97]. They require less solvent and usually a better extraction efficiency (in terms of extraction yield or recovery) is reached comparable to classical SE/LLE. Furthermore, by means of modern MAE and especially SFE approaches, both extraction and clean-up can be performed in a single step [97, 98]. One drawback of PLE/ASE is seen for complex samples, where rather dirty extracts are obtained because of matrix coextraction [91].
3.2. Solid-phase extraction
Solid phase extraction (SPE) is another possibility for direct extraction of liquid samples, in mycotoxin analysis most often used for clean-up and pre-concentration of extracts. Originally, SPE has been performed on broad-range, non-specific stationary phases (reverse-phase, normal-phase, ionexchange, activated carbon etc.), while recently there has been greater emphasis on the use of another type of materials, which enable very selective binding of target molecules and sometimes also higher recoveries [99]. The most popular are immunoaffinity (IA) materials, while molecularly imprinted polymers (MIP) are an emerging, cheaper and very promising alternative [65, 100].
SPE on non-specific materials is still often employed in mycotoxin analysis. This kind of extraction is mainly performed for multiresidual determination of mycotoxins followed by LC-MS/MS [101, 102], where greater selectivity of SPE material would be a limiting element.
IA materials are prepared by binding antibodies specific for a given mycotoxin to a specially activated solid-phase support. Immunoaffinity extraction (IAE) is performed generally for all mycotoxins in very diverse matrices [98, 103-111]. More complex samples require combination of different clean-up procedures (for example LLE, SPE, precipitation of matrix substances) with immunoaffinity columns (IAC) [109, 112]. The IAC are not absolutely selective for individual mycotoxins as also mycotoxin analogues are usually bound to the material. However, when a separation technique is used for the subsequent determination, this does not present a problem [104, 108, 109]. As an additional advantage, IAC for different groups of mycotoxins are commercially available [103, 111]. The main problem concerning clean-up via immunoaffinity columns is the denaturation of the antibodies in the presence of even low concentrations of organic solvents, therefore expenses are rather high [106]. One way to overcome this problem is by extract dilution, although it may cause a loss of sensitivity. The capacity of IAC may also be a problem. Lately, sol-gel immunoaffinity columns have become available. Their use is less time-consuming and less costly; columns have superior storage stability [107].
Usually, the antibodies for IA materials are prepared in vivo by immunization of animals with mycotoxins bound to suitable carrier proteins, but lately IAE on synthetically generated antibodies with specific affinity to mycotoxins has been reported [38, 110].
Another modern SPE with specific material is moleculary imprinted solid phase extraction (MISPE), which enables extraction of a single mycotoxin or a whole group. MIPs are tailor-made synthetic materials with artificial binding sites, able to selectively recognize a target molecule [65, 100]. Preparation of moleculary imprinted polymers is based on polymerization of selected monomers around the template molecule. As a template, target molecule or the so called mimic template can be used, which is usually a structural analogue of the analyte [65, 113]. Preparation procedure of MIPs based on target molecule as the template is problematic with trace analysis, as there may remain residues of the template in the polymer material and therefore leaking of analyte is observed [65, 68]. Additionally, some mycotoxins are too toxic or too expensive for preparation of MIPs [68]. On the other hand, there are also difficulties with selecting the optimal mimic templates. Structurally optimal compounds are not always commercially available as their isolation or preparation could be expensive, especially in the amounts needed for MIPs preparation. Selection of suboptimal mimic template leads to a limited molecular recognition effect [68]. For this reason, many investigations on preparation of MIPs with mimic templates are published: for ZON [66, 67, 100], DON [114], OTA [68] and moniliformin [115]. Usage of MISPE as a clean-up method for the mycotoxin determination increases because of its efficiency in very complex matrices and because of MIPs stability, e.g. in apolar solvents, especially when compared with IA materials [68].
Conventional SPE materials are also used for the clean-up purposes. The most successful is often a combination of different materials [58]; one of such multifunctional clean-up columns is MycoSep [98, 116]. They contain a combination of different sorbents, e.g. charcoal, ion-exchange resins etc., and are designed for easy and rapid handling - extract is obtained within 30 s [39]. The major advantages of these columns are the reduced amount of solvent and avoidance of time-consuming rinsing steps required in single-phase SPE. Other interesting development in the SPE field is primarily the introduction of new materials, e.g. surfactant modified zeolites [117].
All described SPE materials are usually packed in the open cartridges for off-line applications, but also other modes of extraction media are reported: package in short HPLC columns for on-line applications, multiwell extraction plate for high throughput analysis [65, 113], monolithic CIM disks [105] etc. As the sample preparation is frequently the crucial step in the determination of mycotoxins, some on-line extraction procedures, requiring less manipulation of the sample, were shown to be better compared to the off-line approach [24, 58, 118]. Most SPE procedures include the following steps: column conditioning, sample loading, column washing and analyte elution.
Another mode of SPE for solid or semi-solid samples is matrix solid phase dispersion (MSPD), where the sample and sorbent material are mixed homogenously. This mixture is than packed in cartridge and afterwards elution is performed. MSPD on non-selective materials, e.g. silica, phenylsilica, alumina, C8 and C18 has been reported for the determination of AFs in peanuts [119], RALs in fish tissue [28] and trichothecenes in maize flour [120].
Development of powerful separation-identification analytical techniques such as GC-MS and HPLC-MS/MS, which are nowadays routinely used, simplified sample preparation by employing only one single step of LLE or SPE [119, 121-125], but as will be discussed in Section 4, remaining matrix compounds may cause certain problems even with these analytical techniques.
3.3. Other extraction techniques
As the alternative to SPE procedures use of solid phase microextraction (SPME) is reported. This technique employs fibres coated with different stationary phases on which analytes are sorbed from liquid (aqueous) or gaseous samples. Desorption can be performed thermally in combination with GC analysis or with different solvents followed by HPLC analysis. SPME sampling in combination with HPLC analyses was performed for the determination of OTA, CPA, MPA [22, 23, 27, 28, 126, 127]. Another application involving SPME for the mycotoxin determination is the detection of volatile fungal metabolites [82, 83, 85], where also stir bar sorptive extraction (SBSE) can be employed [82]. SBSE uses a coated magnetic stir bar to capture analytes. The extraction phase is mostly PDMS. SBSE has higher recoveries and higher capacity than SPME fibres and therefore additional applications in this field are expected.
3.4. Other sample preparation techniques
Rarely, other sample preparation methods are also used, such as immunofiltration for DON [128] and AFB1 determination [44], diphasic dialysis extraction for PAT determination [129], coacervative extraction for simultaneous extraction and concentration of OTA in wines [130], use of supported liquid membrane for OTA determination [131] and capillary fused silica columns as the traps for the extraction of T-2 toxin from aqueous solutions [132]. Only individual applications are reported so far, but some may find wider application in the future.
Clean-up of the samples has also been reported with the addition of specific chemicals, e. g. zinc acetate [133], hydrogen carbonate and PEG [112], lead hydroxyacetate [134], mostly to precipitate matrix substances, e.g. fatty acids [135]. The majority of these procedures were described for the determination of OTA in different matrices.
4. Analytical methods
The vast majority of chemical analytical methods applied for accurate, selective and sensitive mycotoxin determination in various samples come from the group of separation methods: chromatography, electrophoresis. One of them, high performance liquid chromatography (HPLC) with different detectors is frequently used both for routine analyses and as confirmatory method for novel or screening techniques [35, 45, 51, 58, 71, 81]. For some mycotoxins, e.g. trichothecenes, gas chromatography (GC) is the method more often used [76]. Except for direct mass spectrometric methods, all the other analytical methods used for mycotoxin determination are either immunoassaybased or otherwise fall in the category of direct or indirect screening methods. They were described in details within the Section 2 of the present review.
Similarly to other analytical steps in the mycotoxin determination, the field of separation methods used for this purpose has shown a tremendous development in the last ten years, which can be easily observed from the comparison of analytical reviews of that time [25, 69, 136-140] and in the last few years [16, 19, 39, 141-145]. The great majority of the older and novel methods have detection limits well below the corresponding maximum residue levels in samples [39, 136, 139, 142]. The driving force behind this development is therefore not so much the sensitivity, but the greater simplicity and higher throughput of novel methods. The detector of choice has become the mass spectrometer, preferably even tandem mass spectrometer [16, 141-143]. Judging from the number of publications, fluorimetric detector for HPLC is still very popular due to its sensitivity, selectivity, rather low price and ease of use, although derivatization is necessary for most mycotoxins. Other detectors for HPLC are also used, most notably UV-spectrometric. Good chromatographic separation of analytes is mandatory in combination with all detectors except MS, where some peak overlapping is allowed, therefore the chromatographic conditions will be described only in the second sub-section on LC-FL. As regards the use of other separation methods, GC is often applied for volatile mycotoxins, followed by electrophoretic methods, modern thin-layer chromatography and others [19, 39, 144, 145].
4.1. Liquid chromatography coupled to mass spectrometry (LC-MS)
Liquid chromatography hyphenated to mass spectrometry (LC-MS) or tandem mass spectrometry (LC-MS/MS) has in the last ten years advanced to the status of the reference and definitive method in the field of mycotoxin analysis. The reasons are, among others, the development of efficient electrospray (ESI) and atmospheric pressure chemical ionization (APCI) interfaces for LC-MS coupling, the developments in the field of mass analyzers, and the considerably greater simplicity of use and affordability of tandem mass spectrometers. Probably the greatest merit goes to the ESI and APCI. Before their development, the LC-MS analyses of mycotoxins were performed using thermospray and fast-atom bombardment interfaces, but with significant difficulties [136]. Modern LC-MS instruments with ESI or APCI enable ionization in both positive and negative mode, as well as switching between them in the same chromatographic run [16], which means the best possible detection conditions for all analytes.
Most newly developed LC-MS methods enable analysis of a whole group of similar mycotoxins. For A-trichothecenes, a method using LC-APCI(+)-MS and deuterated T-2 toxin as an internal standard (IS) was developed [146], while in another research with tandem MS [147], ESI in negative mode yielded better results for four B-trichothecenes than APCI in either mode. Electrospray with MS/MS was applied to the semi-quantitative analysis of non-macrocyclic and macrocyclic trichothecenes in samples from indoor environments [148]. Six B-trichothecenes and metabolites were determined by LC-APCI(-)-MS with dexamethasone as an internal standard [149]. Trichothecenes from groups A and B were analyzed using APCI and tandem MS without IS [150] or by ESI and MS/MS with deepoxydeoxynivalenol as IS [151].
Several LC-MS methods have been published for the determination of estrogenic Fusarium mycotoxins - RALs (zearalenone and its metabolites) [142] with a single mass analyzer [109, 140, 152, 153] or tandem MS [28, 91, 140, 154-156]. The ionization was possible both by APCI [28, 109, 152, 154] and ESI [28, 91, 153, 156]. Additional focus was drawn to the determination of the conjugated analytes in samples [157] and of zearalenone metabolites [158]. Matrix compounds can cause a considerable suppression of analyte ionization [28, 39], therefore the use of internal standards is recommended for quantification. A structural analogue zearalanone has been widely used for this purpose [150, 153, 154], although this can be a metabolite as well [155]. Therefore, deuterated internal standards are used as better, albeit more expensive alternative [91, 109, 156, 159]. Additional confusion in the determination of RAL mycotoxins, especially in animal samples, can arise from the presence of structurally related anabolic substance zeranol and its metabolites, which are also ionized by ESI and should be considered in the method [33, 102, 160, 161].
Fumonisins are another group of Fusarium mycotoxins occurring mostly in corn. LC-MS methods employed for their analysis [142] apply single analyzer [162, 163] or ESI(+) with tandem MS [164, 165]. Enniatins and beauvericin, produced by the same fungal genus, have also been determined by LC-MS/MS [142, 166]. Moniliformin, a low molecular weight and highly polar mycotoxin produced by Fusarium species, is efficiently ionized in the negative mode of ESI and APCI [121, 142, 167], but may have to be derivatized in order to achieve retention on reverse-phase LC columns [142]. Alternatively, ion-pairing can be used for this purpose [167]. Methods for the analysis of Fusarium mycotoxins from different chemical groups with LC-MS/MS have been published as well, requiring optimized ionization conditions and switching between positive and negative modes for every group [39, 168, 169]. Use of isotopically labeled IS is preferable [163].
Aflatoxins are mycotoxins produced by Aspergillus fungal species, occurring in various food and feed commodities. However, the published LC-MS methods are relatively uncommon, mainly because of well-established, reliable and sensitive LC methods with fluorescence detectors measuring the natural fluorescence of aflatoxins [142]. Compared to that, there are several LC-MS methods dealing with the precursor sterigmatocystin [142, 170-172]. ESI ionization is applied in most LC-MS methods for aflatoxins [119, 172-174], APCI much less [122], as it seems to be less efficient than the former, except in the case of aflatoxin precursor sterigmatocystin [142]. Atmospheric pressure photoionization (APPI) might be a better alternative to ESI than APCI [142]. Most studies report absence of matrix effects and ion suppression in ionization [142], while Cervino et al. [174] found matrix independent response for AFB2 and AFG2 and strong matrix dependence for other aflatoxins even while using deuterated internal standards, therefore matrix-matched calibration was recommended.
Ochratoxins, especially ochratoxin A (OTA), are another group of harmful mycotoxins for which, due to their natural fluorescence, several well-established and sensitive LC-FL methods exist. Therefore, LC-MS methods aren’t as frequently encountered as for Fusarium mycotoxins. The first methods were published at the beginning of this review’s timespan [138, 175], utilizing ESI ionization. The same interface has featured in most methods up to date, used either in positive [101, 176] or negative mode [127, 173], the former offering a more specific fragmentation pattern [142]. Use of APCI interface results in lower sensitivity due to extensive fragmentation of OTA [142]. There are reports of decreased ionization efficiency because of matrix effects [142], therefore the calibration step is crucial. Nevertheless, some authors report excellent results with external calibration [101, 173], while in other studies ochratoxin B (OTB) was used as an internal standard [127, 142, 177]. Instrument sensitivity for OTB is significantly lower compared to OTA, therefore high concentrations of OTB are needed [127]. Although uncommon by now, the use of deuterated internal standards is preferable [176]. LC-MS has been comparatively more often used for the elucidation of OTA metabolites [142] and products of photodegradation [118].
Published LC-MS methods for the determination of less ubiquitous mycotoxins are not frequently encountered. Alternaria toxins in various fruit samples were determined by LC-MS/MS. ESI in negative mode offered the best sensitivity and specificity compared to ESI(+) and APCI in either mode [178]. Analysis of patulin in apple juice was successfully accomplished with a single mass analyzer in SIM mode [179]. Use of isotopically-labeled internal standard is advisable [180]. In another study, Takino et al. compared APCI and APPI for the same purpose and found that APPI provides lower chemical noise and ionization suppression even in the absence of internal calibration [181]. On the other hand, some authors found LC-MS considerably less sensitive than GC-MS and recommended it for structural confirmation only in spite of the more simple sample preparation [142]. Citrinin can be efficiently ionized by ESI and determined by tandem MS [172]. The most important ergot alkaloids and their isomers of lower toxicity have been determined by ESI-MS/MS [182]. Finally, there is a heterogenous group of mycotoxins produced mainly by Penicillium species, among them cyclopiazonic acid (CPA), mycophenolic acid (MPA) and roquefortin C. A method using LC-ESIMS/MS in SRM mode was employed for CPA in milk samples [183]. Several methods exist for the determination of MPA, which is also a metabolite of an immunosuppression drug [142]. A full-scan and SRM LC-MS/MS method for the analysis of six Penicillium mycotoxins with low detection limits was developed as well [17].
The strength of LC-MS methods lies in the possibility to perform multi-analyte analyses. Due to the highly specific mode of detection in tandem MS (SRM), chromatographic separation is slightly less important and overlapping peaks can be tolerated. Still, the challenges of achieving right chromatographic conditions remain especially in modulating pH and additives in the mobile phase to promote the optimal ionization of analytes in the ion source. Since mycotoxins vary greatly in the polarity, molecular mass etc., optimal ionization can only be accomplished in modern instruments with rapid switching between negative and positive ionization modes required by different chemical classes [136]. Sample preparation and especially extract purification is problematic as it relies heavily on the polarity and functional groups of analytes; simultaneous extraction of different chemical groups without significant matrix carry-over is thus nearly impossible. If no clean-up is applied, significant and unpredictable ionization suppression of different mycotoxins should be expected [123, 124]. Reliable quantification can be achieved only by matrix-matched calibration with model matrix resembling the actual samples as much as possible [124, 184] and also by using isotopically-labeled internal standards whenever possible, preferably one IS per analyte [141, 185, 186]. Structurally analogous internal standards may not compensate for the matrix effect at all, as the ionization efficiency could be different. Although the deuterated IS are preferable, they may still exhibit some unpleasant phenomena, e.g. different extraction recoveries or exchange of deuterium by hydrogen atoms. Stable 13C-labeled IS seem to be the best choice for now [186]. In spite of these drawbacks and potential problems, the number of published multi-analyte LC-MS methods for mycotoxins from different chemical groups and their metabolites is increasing and ranges from “small-size” [17, 150, 172, 187] and “medium-size” multi-analyte methods - above 10 different compounds [151, 169, 188] to “large-size” - above 30 compounds in the same chromatographic run [16, 121, 123, 124, 184]. However, the pioneer LC-UV-MS screening method [189] remains unsurpassed by its 474-compound database of mycotoxins and other fungal metabolites, albeit providing only qualitative data.
4.2. High performance liquid chromatography with fluorescence detection (HPLC-FL)
Fluorescence detection is by its nature highly specific and sensitive. Several well-established, reliable, robust and sensitive LC methods with fluorescence detection exist especially for the determination of mycotoxins with natural fluorescence, e.g. aflatoxins, ochratoxin A, citrinin [142] and many of them have been established as the official AOAC methods. For the others, pre- or postcolumn derivatization with suitable reagents has been widely applied, but has been recently superseded by relative simplicity, but comparative specificity of LC-MS methods after the lower cost has made these instruments available to most laboratories. Nevertheless, LC-FL might still be superior in the area of quantitative determination, where the influence of matrix is negligible compared to possible problems with LC-MS quantification [141].
In spite of its specificity for the fluorescing compounds, they have to be well separated on the chromatographic column to enable a reliable quantification. Usually, a reverse-phase stationary phase is used, e.g. C18. Base deactivation additionally improves peak shape for polar mycotoxins with carboxylic groups: citrinin, OTA, some fumonisins [138]. Ion-pair HPLC has also been applied for the polar toxins, using tetrabutylammonium hydroxide [138]. Mobile phase should be composed of acidic aqueous phase (acetic acid, trifluoroacetic acid, acidic buffer) to prevent ionization of carboxylic groups, and gradient with methanol or acetonitrile is preferred [138].
Ochratoxin A is probably the mycotoxin most often determined by HPLC-FL. Most authors use the octadecylsilica stationary phase and isocratic mobile phase composed of acetonitrile and water acidified with acetic acid (approx. 1:1) [99, 112, 190-193]. Retention time for OTA is below 15 min under these conditions. Alternatively, a phenylhexyl column is chosen [31]; methanol is sometimes used instead of acetonitrile [194] or a mixture of both [195]. Mobile phase is usually slightly acidified with acids, but pH may not be stable; therefore some authors prefer the use of acidic buffers [195, 196]. Medina et al. [134] published a thorough investigation on suitability of various isocratic mixtures for OTA separation and found methanol unsuitable. Peak for OTA was wide and tailing if pH of mobile phase was above 3.5; it was therefore necessary to use phosphoric acid for acidification [134]. Fluorescence is usually measured at λex 330-334 nm and λem 460-464 nm [99, 112, 134, 190- 194], but some authors use lower excitation wavelengths 225 nm [195] or 247 nm [196] and different emission wavelength of 480 nm [196]. There is little variation in the chromatographic conditions, but some innovative approaches include automatization of the analytical procedure [197], and inclusion of cyclodextrins [198] or post-column ammoniation [199] to enhance fluorescence. Most publications, however, focus on the sample pretreatment methods to minimize the matrix interferences. Besides the immunoaffinity clean-up [29, 99, 112, 190, 192, 193], conventional SPE is used [31, 112, 194, 196]. Other sample preparation approaches are sometimes applied, such as on-line SPE [118], extract cleaning with lead hydroxyacetate [134], solid-phase microextraction [126, 191] and coacervative extraction [130]. In spite of clean-up, coeluting substances may appear in the chromatograms when dealing with complicated matrix [190]. In such cases, confirmation of OTA identity is accomplished by derivatization to fluorescing methyl-OTA with longer retention times [112, 190, 192, 194] or with LCMS [194]. Practically all studies apply external calibration or sometimes standard addition method [192], internal standards being rarely used, e.g. diflunisal [200]. Compared to the vast number of articles dealing with HPLC-FL determination of OTA in various matrices, there are only rare publications dealing with similar compounds, e.g. ochratoxin B and metabolite ochratoxin α [201, 202].
Aflatoxins possess natural fluorescence as well, at least AFB2 and AFG2, while AFB1 and AFG1 (see Figure 1) have to be derivatized to enhance their fluorescence [13, 103, 108, 111, 119, 203, 204]. The most usual is derivatization to their hemiacetal form prior to chromatographic separation, using trifluoroacetic acid [13, 103, 108, 119, 203, 204], although post-column derivatization with pyridinium hydrobromide perbromide has been applied as well [111]. Fluorescence detection is accomplished at λex 360-370 nm and λem 418-425 nm [13, 14, 108] or 435-440 nm [103, 111, 119, 203, 205]. The usual chromatographic conditions are reverse-phase (C18) column and isocratic mobile phase consisting of 20-50 % organic solvent, usually a mixture of methanol and acetonitrile, and water [13, 103, 108, 119, 203, 205]. Some authors use aqueous phase acidified with phosphoric acid [14, 111]. The determination is frequently performed together with OTA determination. A step-wise gradient is usually applied, switching to isocratic mobile phase suitable for OTA elution after aflatoxins have been eluted (12-20 min) [13, 14, 103, 111, 203]. Considerable accent has been put on sample preparation rather than chromatographic separation, which seems to be non-problematic. Besides immunoaffinity clean-up [14, 29, 103, 108, 111, 203-205] there are some new approaches, e.g. automation of this procedure [111], use of immunoaffinity monolithic discs (CIM) for on-line extraction [105], SPE on silica columns [206] or matrix solid-phase dispersion (MSPD) extraction [119]. Airborne aflatoxins and OTA have been collected in water by passing air through it, followed by immunoaffinity clean-up [14] or sampled on microfibre filter subsequently extracted with organic solvents [13]. Again, there are relatively few methods published for the determination of the metabolite aflatoxin M1 [29, 205].
Fusarium mycotoxins, important pollutants in various foodstuffs, do not fluoresce and have to be derivatized, usually before chromatographic separation. With fumonisins, the reaction site is the primary amino group and the reagent most often used is o-phthaldialdehyde (OPA) with 2- mercaptoethanol [35, 39, 51, 139, 207-212]. The resulting derivate is highly fluorescing (λex 335 nm, λem 440 nm), but stable only for 4 min [39, 138, 139]. As an alternative to 2-mercaptoethanol as reaction partner for OPA, N-acetyl-cystein has been identified as the most suitable [211]. Various other derivatization reagents have been tested: fluorescamine, naphthalene-2,3-dicarboxaldehyde (NDA), 4-fluoro-7-nitrobenzofurazan (NBD-F), dansyl chloride, 9-fluorenylmethyl chloroformate (FMOC), 6-amino-quinolyl N-hydroxysuccinimidylcarbamate, fluorescein isothiocyanate (FITC) with different stabilities and sensitivity of detection [39, 138, 139]. NDA derivatives appear to be the most promising alternative [212]. The column for the chromatographic separation of the derivatized fumonisins is reverse-phase (C18 or phenylhexyl), but as the compounds still have four carboxylic groups, strongly acidic mobile phase is needed to obtain good peak shape [138]. Usual mixtures are composed of methanol or acetonitrile and acidic buffer at pH approx. 3.5 [35, 207, 208], isocratic or linear gradient. Native fumonisins can be separated on ion-pair column and derivatized postcolumnally with OPA and N-acetyl-cystein [139]. Immunoaffinity [139, 208, 209, 212] or SPE on C18 and SAX columns [139, 207, 210] are applied prior to HPLC. Methods for the metabolites, e.g. aminopentol-1, are seldom published, but use essentially the same procedure [207].
Trichothecenes, another important group of Fusarium mycotoxins, are amenable to GC analysis with and without derivatization, therefore this has been the most frequently applied method. The latter is applicable only to less polar compounds, e.g. T-2 toxin, 4,15-diacetoxyscirpenol [136]. However, some methods employing derivatization to fluorescing products and subsequent HPLC-FL analysis have also been published. One of the first was a post-column degradation of DON and nivalenol to formaldehyde by sodium hydroxide, followed by the formation of fluorescent derivate from reaction with acetoacetate and ammonium acetate [39, 136]. Method was later extended to other type-B trichothecenes [39], but is quite experimentally demanding [136]. Coumarin-3-carbonyl chloride is the most often used reagent for off-line derivatization of type-A and B trichothecenes prior to HPLC [209, 213-215]. The derivatives are then separated on reverse-phase columns using a mixture of acetonitrile and water acidified with acetic acid. High percentages of acetonitrile should be present (> 60 %) [209, 213, 215]. Methanol may be used as well [214]. Fluorescence detection is performed at λex 292 nm and λem 425 nm. Excess reagent peaks might interfere with the detection, so it is advisable to perform clean-up after derivatization [213]. Alternative derivatizing reagents include 1-anthroylnitrile, where products are separated under similar conditions as above and detected at λex 381 nm and λem 470 nm [106]; zirconyl nitrate and ethylenediamine, although in this method no HPLC separation was performed [72]; pyrene-1-carbonyl cyanide for T-2 toxin in the presence of cyclodextrins [198]. In spite of these efforts, HPLC-UV [215] and more recently LC-MS [39] are more frequently applied because of relative simplicity and comparable or better sensitivity.
Zearalenone and zearalenols are compounds with native fluorescence. Common HPLC-FL methods thus include separation on C18 column with mobile phase with high percentage of organic solvent and detection at λex 270-280 nm and λem 440 nm or 452 nm [39, 45, 93, 116, 209]. Fluorescence can be further enhanced by cyclodextrins [189]. The main emphasis of method development is on the extraction and clean-up of samples before the analysis, mostly using immunoaffinity columns [14, 116, 209]. Alternative approach was accelerated solvent extraction without further clean-up [93]. While HPLC-FL was still the method of choice for zearalenone seven years ago [140], it has now been superseded by LC-MS methods [39, 145].
Citrinin, a mycotoxin produced by different fungal genera, has acidic properties and is in its nondissociated form a fluorescing compound (λex 320-340 nm and λem 495-512 nm) [19]. Chromatographic separation has been achieved on normal-phase columns, reverse-phase and reversephase ion-pair stationary phase [19, 138]. The first method suffers from low retention time reproducibility [19]. In reverse-phase chromatography, mobile phase has to be acidic, while acetonitrile is preferable to methanol to achieve better sensitivity. It is quite difficult to achieve good peak shape for citrinin [19]. Thus, ion-pair RP with tetrabutylammonium hydroxide or phosphate yields the best chromatographic results, but the eluate has to be acidified prior to detection in order to enhance the sensitivity, as the anionic form of citrinin does not fluoresce [19, 138]. Due to the similar chromatographic behavior and fluorescent properties, as well as frequent occurrence in the same type of samples, methods for simultaneous determination of citrinin and ochratoxin A are sometimes encountered [138, 216]. Ergot alkaloids are also sometimes analyzed by HPLC-FL. The published methods are dealing only with determination of ergovaline in various matrices [217-219]. This toxin has the native fluorescence (λex 250 nm, λem 420 nm) and can be separated on reverse-phase column using isocratic mobile phase. Ergotamine may be used as an internal standard [218, 219].
In spite of some obvious advantages of fluorescence detection, such as excellent specificity, low limits of detection, robustness and independence from matrix, HPLC-FL methods have been mostly swept away by LC-MS. The main advantage of the later is in its identification power and possibility to perform determination of multiple analytes from different chemical groups in the same run. Thus, it is expected that the development of methods employing fluorescent detection of mycotoxins has at least temporarily stopped.
4.3. High performance liquid chromatography with other types of detection
Compared to the mass spectrometric and fluorescence detection, all other detections available in HPLC are seldom used in mycotoxin analysis. The reasons might be in higher limits of detection unsuitable for trace amounts of the determined substances, and lack of specificity for some of detectors.
A large database of mass spectrometric and UV absorption data for 474 different mycotoxins and other fungal metabolites has been published by Nielsen and Smedsgaard [189]. As can be seen from it, most of the important mycotoxins presented in this review do not absorb in the UV part of spectra at all (e.g. most of trichothecenes, enniatins, fumonisins) or absorb only at rather non-specific wavelengths 200-225 nm (e.g. DON and its derivatives, ochratoxins, some zearalenone analogues) [189].
HPLC methods for the determination of Fusarium mycotoxins using UV detection have been published for deoxynivalenol (λ 218 nm) [220-222]; for nivalenol, DON and some of its derivatives [223] or for several type-B trichothecenes (diode-array, λ 221 nm) [215]. In the latter method, UV detection gave better results than FLD of coumarin-3-carbonyl derivatives [215]. Method with simultaneous UV and FL detection of trichothecenes type-B, OTA, zearalenone and citrinin was also developed [216]. Better sensitivity for DON and other type-B trichothecenes can be achieved by electrochemical detection in reductive mode or by derivatization with p-nitrobenzoyl chloride and subsequent UV detection at 254 nm [136]. Zearalenone and its metabolites are also amenable to UV detection, e.g. at λ 236 nm [209, 224]. Alternative mode of detection is amperometric [71]. Fumonisins do not absorb UV radiation, but evaporative light scattering detector (ELSD) has been applied in their analysis [138, 225].
Ochratoxin A, routinely analyzed by HPLC-FL, can also be detected with UV (photodiode-array) detector at 333 nm. Its presence was confirmed by peak shift after esterification with BF3 [226].
Patulin is a mycotoxin perfectly amenable to HPLC (reverse-phase) with UV detection because of its strong UV absorption - λmax 276 nm [25, 189]. Therefore, some methods have been published [25, 227], but particular care has to be taken to separate patulin from 5-hydroxymethylfurfural, a coeluting compound of non-fungal origin commonly appearing in deteriorating fruit juices [25, 228].
Cyclopiazonic acid exhibits strong UV absorption at cca. 280 nm, therefore some methods for its determination by HPLC-UV (photodiode-array) have been published. Separation and peak shape might be problematic because of the acidic nature of the analyte, but were nevertheless plausible on C18 column using a gradient of solvent containing ZnSO4[20]. Another possibility is an aminopropyl-bonded silica as a stationary phase and mobile phase, composed of acetonitrile and ammonium acetate. In this case separation is accomplished by mixed ion-exchange and reverse-phase mechanisms [27, 229].
LC-UV methods for other mycotoxins are not frequently published. Mycophenolic acid in cheese was determined on aminopropyl-silica column with mobile phase at neutral pH and 80 % of organic solvent using a previous extraction with solid-phase microextraction. Detection wavelength was 254 nm [23]. Roquefortine C in cheese was analyzed by on-line extraction and separation on C18 column, λ 330 nm [24]. Alternaria mycotoxins can be separated on reverse-phase column with UV detection at 240-260 nm [230]. Citrinin exhibits maximal UV absorbance at non-specific 216 nm. Although LC-UV methods exist for its determination, other methods are preferable [19].
4.4. Gas chromatography (GC)
Modern gas chromatography combines superior separation on the capillary columns with a variety of general and specific detectors. The obvious disadvantage when compared to LC is the fact that only thermally stable and volatile analytes can be analyzed, although this problem can partially be solved by derivatization. However, as mycotoxins are generally speaking rather large and polar molecules, they are better amenable to LC and GC methods are not common.
Of the published GC methods prevail those developed for trichothecenes and other Fusarium toxins [39, 136, 138]. Onji et al. [135] report on analysis of native compounds (zearalenone, DON, T-2 toxin and others) using cool on-column injection and there are other reports on direct analysis [136, 138], although adsorption of more polar mycotoxins to column has been observed [136]. An obvious advantage is the combination with MS detection, as the identity of compounds can be confirmed [135], which is especially important in the analysis of metabolites [136]. Direct analysis of T-2 toxin using a GC-GC tandem system for sequential extraction and separation was also accomplished [132]. Most researchers, however, prefer to derivatize mycotoxins to enhance their volatility and decrease polarity. In trichothecenes, hydroxyl groups are usually derivatized to trimethylsilyl (TMS) [136, 231, 232] or trifluoroacetyl (TFA) derivatives [136, 233]. Other reagents yielding pentafluoropropionyl (PFP), heptafluorobutyryl (HFB), trimethylchlorosilyl (TMCS) derivatives and commercial mixtures in various combinations are also used [39, 136, 234]. Introduction of halogens into the molecule renders it detectable by electron capture detector (ECD), which is highly selective and sensitive [39, 136, 223]. Other detectors for any type of derivatives or native compounds include flame ionization detector (FID) [232] and most often mass spectrometer (MS) with electron ionization (EI) [135, 231, 232] or chemical ionization [233]. Fluoroacyl derivatives are amenable to negative chemical ionization (NICI) as well [136]. However, unknown trichothecenes or metabolites are difficult to identify from their MS spectra if they are derivatized [136, 235]. Other Fusarium toxins may be derivatized and analyzed by roughly the same procedure, using GC-MS: e.g. fusaproliferin [234], RALs [39, 235]. GC methods for fumonisins have also been published. The protocol included hydrolysis of acidic side chains and their re-esterification or acylation of the remaining aminopolyol. Derivatives were subjected to GC-MS analysis, but none of the approaches was satisfactory in terms of compound identification [39, 138]. Analysis of TMS-fumonisins was also developed more than ten years ago [138], but no further attempts were seen in this direction.
Published methods dealing with GC analysis of other than Fusarium mycotoxins are rather scarce. A method for ochratoxin A was developed, employing TMS-TFA derivatization and GC-MS, but it was deemed not suitable for routine quantitation [226]. Some methods for patulin were published, mostly using derivatization to TMS-patulin and ECD or MS detection [25, 236]. In another publication, acylation after diphasic dialysis extraction and GC-MS were proposed [129]. Direct GCMS of underivatized patulin was possible using on-column injection [25]. Internal standards nitrobenzene [129] or 13C-labeled patulin [180] were proposed. Finally, a GC-MS method without previous derivatization was proposed for citrinin [237].
4.5. Thin-layer chromatography (TLC) and other chromatographic methods
Thin-layer chromatography (TLC) is a low-cost, rapid analytical technique, yielding qualitative or semi-quantitative estimations by visual inspection, but with densitometric measurements also reliable quantitative results [39, 69, 145]. These methods were mostly developed before the great development and affordability of HPLC and LC-MS instruments and some were established as the official AOAC methods. Their many advantages include suitability for crude extract analysis, a wide choice of stationary and mobile phases, as well as an array of spraying agents used for the detection [69]. In spite of that, TLC methods are now rarely used for other than screening purposes. Nevertheless, there are some more recent publications showing the interesting possibilities offered by this technique.
A TLC method with previous immunoaffinity clean-up and densitometric quantification has been developed for aflatoxins in food samples. Advantages of the method include fast sample processing and limits of quantification lower than established by regulatory organs [238]. Overpressured-layer chromatography [239], two-dimensional TLC and high performance TLC (HPTLC) are other possibilities for efficient separation and determination of aflatoxins [240]. Sterigmatocystin, a precursor to aflatoxins, has been determined on amino-derivatized HPTLC plate and reagent-free (only heating) densitometric detection [241].
While aflatoxins are naturally fluorescent compounds and their detection in TLC is not problematic, trichothecenes require post-development visualization with spraying reagents: p-anisaldehyde, 4-pnitrobenzylpyridine [39], and aluminum chloride for DON [242]. Recent methods include quantitative determination of DON and T-2 toxin in cereals by high performance TLC [242, 243]. Fumonisins have also been determined in corn using reverse-phase TLC plate, spraying with fluorescamine in borate buffer/acetonitrile mixture, and densitometric detection [244]. In contrast to older methods [39], this application has had acceptable limits of detection for routine analysis [244]. In another method, fumonisin B1 has been analyzed by TLC after clean-up on ion-exchange column. The spraying reagent was p-anisaldehyde, detection was by laser-scanning densitometry [245]. Zearalenone emits fluorescent light upon excitation. Quantitative methods are based on TLC with densitometry [246] or HPTLC [247]. Moniliformin, a low molecular weight Fusarium mycotoxin, can also be determined by TLC and visualized by 3-methyl-2-benzothiazolinone hydrazone (MBTH) or 2,4-dinitrophenylhydrazine with low limits of detection [39].
TLC determination of ochratoxin A (native fluorescence-densitometric detection) was compared to different HPLC methods. Recoveries for the TLC method were lower than average of HPLC methods [99]. There hasn’t been much development in recent years for the TLC methods for patulin. Generally, the analyte has to be visualized by MBTH or ammonia fumes. Separation from the commonly present contaminant in apple juice, 5-hydroxymethylfurfural, was often problematic [25]. The situation with citrinin is similar, as the TLC methods used before the expansion of HPLC-based protocols were generally not sensitive enough. Citrinin was visualized by exposure to aluminum chloride, pyridine or acetic anhydride. Silica plates had to be impregnated with acid to achieve symmetrical spots [19].
Other chromatographic techniques besides HPLC, GC and TLC are a rarity in mycotoxin analysis. Moniliformin from maize plants was separated by hydrophilic interaction chromatography (HILIC). Detection was accomplished by UV detector at 229 nm and with negative-mode electrospray mass spectrometer (ESI(-)-MS) [248]. Combination stationary phases operating both through RPhydrophobic and hydrophilic interactions are expected to find broader application in the future due to good retention of neutral, acidic and basic compounds. They are especially well suited for multi-toxin LC-MS/MS methods where a great number of chemically diverse mycotoxins should be separated in a single chromatographic run [249]. Trichothecene analyses in fungal cultures were performed by supercritical fluid chromatography (SFC) and electron ionization MS detection (EI MS) in the heyday of this technique, but there were no further developments [136]. High-speed counter-current chromatography (HSCCC) has been used as a preparative rather than analytical technique for isolation of altertoxin I from Alternaria species [250] or some novel fungal compounds from Penicillium species [251].
4.6. Capillary electrophoretic (CE) methods
In spite of the great separation power and versatility of capillary electrophoretic (CE) techniques, they have never gained such popularity as HPLC, although the same analytes can be determined with CE and the same detectors can be used. Possible explanation might be in better detection limits and greater user-friendliness of HPLC methods.
Achieving low enough detection limits might present a serious problem in CE, therefore the mycotoxins for which CE methods have been developed are mostly those for which fluorescence detection (FL) can be used. Ochratoxin A was determined by CE-laser induced fluorescence (LIF) in human serum [55], food samples after immunoaffinity clean-up [252], and in wine after combined extraction with SPE and supported liquid membrane [131]. Capillary zone electrophoresis (CZE) coupled to LIF was applied to OTA analysis in house dust [253] and human serum [254]. Aflatoxins in animal feed were determined by micellar electrokinetic capillary chromatography and FL [70]. Cyclodextrins were applied to enhance native zearalenone fluorescence in CE-LIF analysis [255], and for the same purpose in CE-LIF determination of T-2 toxin, which was previously derivatized with pyrene-1-carbonyl cyanide [198]. Another detector commonly used in CE is UV-spectrometric. Methods have been developed for determination of patulin in apple juice, employing micellar electrokinetic capillary chromatography (MEKC) [256] or microemulsion electrokinetic chromatography (MEEKC) [257] and UV detection at 276 nm. Limits of detection were in the μg/L range, which is satisfactory for routine analysis. In contrast to the CE-FL methods for ochratoxin A mentioned above, UV detection of OTA after CE separation has given quantification limits that were too high for the analysis of real samples [195].
4.7. Other analytical methods
Applications of analytical methods other than separation or immunoaffinity-based techniques, e.g. to study the electrochemical behavior of mycotoxins [258], are sometimes encountered, although the applied concentrations are too high to be applicable to trace analysis. The only other technique used in mycotoxin analysis in the food samples is ion mobility spectrometry (IMS). It has been applied to determination of aflatoxins in pistachios, the ionization method was corona discharge [259]. Another example is the determination of zearalenone in maize samples by high-field asymmetric waveform ion mobility spectrometry - mass spectrometry (FAIMS-MS) with electrospray ionization [260]. Both methods achieved good limits of detection.
5. Determination of mycotoxins in various matrices
After the general overview of the screening, sample preparation, clean-up and analytical techniques, more detail on the published methods for the individual mycotoxins in various matrices is presented in Table 2.
Table 2. Methodology for determination of mycotoxins in different matrices.
The main considerations when choosing the appropriate set of techniques suitable for the mycotoxin and the sample are the following: homogeneity of the sample is achieved with greater ease with liquid samples, to which also direct solid-phase extraction techniques may apply. For solid samples, however, the sample comminution and homogenization is the most crucial step to assure the representative sample. Extraction is then almost invariably performed by solvents, using either classical (Soxhlet) or novel approaches (PLE, SFE, MAE). Decision to clean-up the crude extract depends mainly on the type of matrix and presence of potentially interfering compounds, e.g. sugars, fatty acids etc., as well as detection mode. Finally, the choice of the analytical method depends mainly on the chemical characteristics of the mycotoxins.
6. Pitfalls of mycotoxin analysis
Mycotoxin analysis presents several problems for the analytical chemistry, many of which have already been at least in part addressed in the previous sections. There is a formidable diversity of chemical structures and associated physico-chemical properties, and on the other hand a vast array of samples containing only trace amounts of these compounds, but a lot more of interfering substances. As we are dealing with an opponent that has to be respected, we feel it is necessary to reemphasize and elaborate on certain aspects of mycotoxin analysis that are exceedingly important for the final result. The accuracy of the latter is, as ever, dependent on the accuracy of the steps taken to obtain it [283].
6.1. Metabolites and other transformation products
Although grouped in the same subsection, there are several separate problems that have to be addressed here. The first question to be asked is: do we really determine all of the mycotoxins present in our sample by the chosen analytical method? Apart from the loss of the analytes during the sample preparation step and inaccurate analytical determination, which will be discussed later, the problem frequently arises from the fact that we are looking only for the free mycotoxins in the sample and forget about their various conjugates, which nevertheless cause the same or similar physiological impact when ingested. Conjugated mycotoxins result from the infected plant metabolism of the mycotoxins excreted by the infecting fungi [16, 158]. Our knowledge of the mycotoxin identity stems mainly from their isolation from the laboratory-grown fungal cultures [189], but metabolites formed in infected plants have been taken in consideration only recently and nicknamed “masked mycotoxins” [16, 158]. As an example, at least 17 different metabolites/conjugates of zearalenone have been identified in the model plant [158]. Detection and identification of novel metabolites in a complex mixture is a hard task, therefore protocols and computer algorithms are developed to aid in this process [284]. Important conjugates in terms of frequent occurrence have been found for zearalenone [16, 157], ochratoxin A, and DON in wheat and maize [16]. Processing of foodstuffs, e.g. cooking and frying, is another source of mycotoxin conjugation [16] or transformation [9, 19, 285]. Fumonisin B1 was thus shown to react with sugars, amino acids and proteins during cooking [16], while citrinin was transformed into more toxic product citrinin H1 [19] and less toxic citrinin H2 [19, 285], depending on the temperature of heating. Finally, mycotoxins can be degraded upon exposure to light, yielding products of unknown toxicity. This has been shown for roquefortine C in cheese [24] and for ochratoxin A in wines [118].
Determination of mycotoxins in food samples derived from animals (meat, dairy products, eggs) presents other challenges. Some mycotoxins exhibit very low carry-over from feed to animal products: DON, fumonisins, zearalenone [3, 282], e.g. the transmission rate of DON to eggs was shown to be 15- 29·103 :1 [274]. Nevertheless, some metabolites have been identified, e.g. deepoxy-DON in pig urine [149] or aminopentol-1, a fumonisin B1 metabolite, in swine liver [207]. Zearalenone is metabolized primarily to α-zearalenol and β-zearalenol, but further into zeranol and taleranol [155]. Subsequent metabolites include also α-zearalanol, β-zearalanol and possibly zearalanone [109, 286], although the latter couldn’t be detected in all studies [155]. Things get further complicated with zearalanone, because it has been used (and still is) as a very satisfactory internal standard in the mass spectrometric determination of zearalenone and its first metabolites [153, 154] before it was known that it occurs in nature as well. Finally, a banned anabolic substance for farm animals zeranol also yields metabolites taleranol, zearalanone and zearalenols [160].
Ergovaline, an Ergot mycotoxin, was shown to be excreted into milk only in a limited amount [219], similarly to ochratoxin A, which accumulates mainly in blood, liver and kidney, but not in muscles [3]. However, OTA is excreted in urine as a conjugate with glucuronic acid [287].
One of the most important metabolites to be determined in animal samples is aflatoxin M1, occurring in milk and dairy products [29, 63, 205, 276-278]. Carry-over of aflatoxins has been estimated to 1-2 % [3]. Unchanged aflatoxin B1 was excreted in eggs at transmission rate 5000:1 [275]. In meat and meat products, other metabolites besides M1 have been detected: aflatoxin M2 and P1, aflatoxicol [108].
Besides food and feed, other samples are of importance in mycotoxin analysis. Environmental samples are a potential source of contamination of plants and drinking water, but degradation products of mycotoxins may occur as well. In the survey of various real-life aqueous samples for zearalenone, it was present in drainage water from the infested field, but none of its metabolites were detected [156]. In another research, degradation of zearalenone and OTA was studied in agricultural soils. Half-lives were about 10 days for ZON and below 1 day for OTA, but no degradation products were identified [32]. Human biological fluids (blood, urine) are monitored for the presence of mycotoxins and their metabolites for the purpose of assessing and estimating the exposure of population [283]. While native compounds are most frequently determined, metabolites - if known - can usually be better biomarkers of exposure [279, 283, 287].
6.2. Choice of the representative sample
Sampling is an extremely important part of the overall analytical procedure in general, but even more so in the determination of mycotoxins. The reason is a highly heterogeneous distribution of analytes (“hot spots”) in the lot of food/feed commodities. Moreover, the sampling stage of the analysis directly influences the costs of the control procedure [283]. In spite of the extreme importance of sampling, the laboratory performing the analysis usually has no influence on it [39]. It has been estimated that the coefficient of variation (CV) in the determination of aflatoxin concentration in peanuts is between 60 % and 120 % for sampling, and only up to 20 % for subsampling and up to 10 % for the analysis [283, 288]. Interestingly, DON determination in wheat showed lower CVs for sampling than for sample preparation in spite of small sample size (< 0.5 kg) [289]. It would therefore seem that sampling error might depend both on the type of sample and mycotoxin. The effects of improper sampling and subsequent inaccurate determination are either that unsuitable lots are put on the market (false negatives: risk for the consumers) or harmless lots withdrawn (false positives), presenting a financial burden for the producers [283, 288].
Standardization of sampling procedures is therefore absolutely necessary to minimize the occurrence of both false positives and negatives [288]. On the other hand, the sampling procedure should also be fast, cost-effective and easy to apply [290]. Although most samples needed to be analyzed are from the food/feed lots waiting to be released on the market, sampling should not be forgotten elsewhere, e.g. in the research of mycotoxin contamination of grain from field to the consumer, where proper sampling procedures in each step also affect the final result [291]. Some strategies to decrease the determination error associated with the sampling are reviewed below.
The simplest method to reduce sampling error is by increasing the sample size [288], although this may be impractical. Another approach is to locate a considerable number of sites (>100) in the lot and form an aggregate representative sample from these partial samples [283]. This approach, however, requires a statistically correct sampling plan to cover the whole lot [290]. Another problem with big samples is their subsequent homogenization and comminution in order to obtain a representative laboratory subsample. It has been shown that slurry mixing of samples results in lower CVs than dry milling [292], although the obtained slurry presents problems for the disposal after the analysis [290].
Some novel methods aim to avoid these difficulties by performing a more-or-less “sampling-free” screening. Lots deemed positive should be sampled and analyzed to confirm the result, but the proposed approach nevertheless considerably decreases time and costs.
An automated sampling system DiscoveryCERT FQS™ developed for the extraction and collection of chemical hazard contaminants from cargo pallets has been implemented to the sampling of corn, pistachio and wheat samples for the mycotoxin analysis. Samples of up to 50 kg could be loaded in the system [290]. The system uses pressure cycling, vibration, air jetting, air blasts etc. for the agitation and release of vapor and particles from the sample material, which are then collected on glass fiber filters with a sampling lance. The filter materials are ground or made into aqueous slurry for subsequent analysis. The results correlate rather well with the concentration of mycotoxins determined by the standard sampling approach [290].
Electronic noses detecting volatile compounds produced by fungi have been applied not only to detect fungal contamination of grain [84, 85, 87] and apples [86], but also to predict mycotoxin concentration [86, 87]. Similarly, solid-phase microextraction or headspace withdrawal with subsequent GC-MS analysis can be used for the same purpose [82, 84-87]. These methods can be performed on the whole lot (or rather over it) of food/feed commodity.
As regards subsampling, the usual procedure is mixing and grinding of the sample to ensure homogenization. However, it has been shown that the distribution of the mycotoxin contamination is related to particle size [77, 283]. Grinding procedures yielding finer particles, such as slurry mixing [292], are therefore preferable, as well as analysis of the most representative ground fraction separated by sieving [77]. Larger subsamples are also associated with lower CVs [283].
6.3. Sample preparation bias
The next step in the analytical determination of mycotoxins is the preparation of the samples for the subsequent analysis, which is also prone to various errors. One dubious advantage may be that this step is usually performed by highly trained laboratory staff, which can more readily observe the possible problems. The well known problems with sample preparation are insufficient purification and consequent interferences during the analysis; introduction of artifacts because of too harsh extraction conditions (e.g. in Soxhlet apparatus); loss of analyte and incorrect estimation of its recovery. A good approach to avoid especially the last problem is the introduction of internal standard in the sample before the extraction and clean-up. Other critical points of sample preparation are highlighted below.
Generally, sample preparation consists of extraction and clean-up step, which may be performed together in some approaches, e.g. if the extraction is selective enough. On the other hand, the raw extract (i.e. without purification) can already yield satisfactory results in immunoassays [39, 57]. Several publications have shown that the analysis of raw extracts can be successfully accomplished by LC-MS [123, 124, 142, 152]. In spite of these reports, coextracted matrix compounds may present a problem even for selective MS detection. Even in the case of good chromatographic separation from interferences, which are “visible” only in one-stage mass analysis, there remains a greater chance of pronounced matrix effects - ionization suppression in LC-MS interface with raw extracts, leading to erroneous quantification [91, 142, 146]. Use of internal standards is therefore practically mandatory in such experiments [39, 142, 146], but even this may not always suffice [174, 186].
Raw extracts are usually obtained by solvent extraction of the sample material. The choice of the solvent or a mixture of solvents is of great importance here, having a direct impact on the recovery of mycotoxins from the matrix. Although the mixture of acetonitrile or methanol with water is most frequently applied, it is not equally suitable for all mycotoxins from every matrix [142, 224, 267]. Especially mycotoxins with acidic functional groups (fumonisins, ochratoxin A) require an addition of acidic modifier to enhance the recoveries, while some samples, e.g. animal tissues, respond better to polar organic solvents [39, 142]. Various substances are added to the sample to precipitate bulk matrix components [134] or enhance mycotoxin extraction [142]. To speed up the extraction step, several approaches have been devised: shaking, blending, microwave assisted extraction (MAE) [97], pressurized liquid extraction/accelerated solvent extraction [39, 142], but especially in the latter higher levels of coextracted matrix compounds have been observed [142]. Additional critical steps are phase separation [267] and defatting with partitioning into nonpolar organic solvent, which is needed for extracts from samples with high lipid content [142]. Both may lead to further analyte loss. Another consideration is about the conjugated mycotoxins in some matrices, e.g. urine, which are better off hydrolyzed prior to extraction [142]. Finally, a word of caution regarding the estimation of recoveries and method calibration using the spiked samples. As it has been shown for many matrices, mycotoxins added to the sample are not so strongly embedded in the matrix as the native ones, therefore longer extraction times are needed for the naturally contaminated samples [39, 138].
The most frequently used approach for the clean-up of raw extracts is the use of immunoaffinity columns (IAC), which show high selectivity for chemically similar group of mycotoxins [39, 142, 146] or to a certain extent even for single compounds [38]. If single compounds are the target, crossreactivity of IA material may present a serious problem with non-specific analytical method [36]. The purification of extracts by IAC for chemically similar group is very efficient and is thus especially suitable in combination with non-specific analytical techniques, but is considered by many researchers an analytical “overkill” if used prior to highly selective LC-MS/MS analysis [142]. In the era of multitoxin methods performed by LC-MS/MS [16, 121, 123, 124, 184], compound-specific purification of extracts even presents an obstacle [142]. Other drawbacks of IAC include rather low capacity [224], significant loss of some analytes (e.g. fumonisins) [264] and incompatibility of columns with some matrices [265]. The comparably higher costs of IA columns should also be considered.
Moleculary imprinted polymers (MIPs) are increasingly popular as an alternative to IAC for selective clean-up of extracts [65, 66]. While being cheaper, reusable and less sensitive to harsh conditions than IAC, they have problems of their own: non-specific binding, lower reproducibility, potential contamination of the extract by template bleeding [68]. The commercial availability is currently low, although this is expected to change in the future [65, 293].
Conventional solid-phase extraction (SPE) is the mainstay of extract purification for mycotoxin analysis, especially the MycoSep line of multifunctional cartridges, designed for easy and rapid handling [39]. However, they have been shown to have low recoveries for zearalenone [142]. A variety of SPE cartridges packed with a single sorbent are used as well: strong anion-exchange resins (SAX), aminopropyl, carbon materials, polymeric resins, reverse-phase (C18) etc. The choice of sorbent depends on the nature of the analyte and the sample, but also on the analytical method used [39, 112, 142]. As an example: SAX cartridges are especially suitable for acidic mycotoxins, e.g. fumonisins, but their hydrolyzed metabolites are not retained [39]. On the other hand, RP sorbent material is rather unspecific, but is thus advantageous in combination with multitoxin LC-MS/MS analyses [142, 146]. One additional advantage of SPE is its ease of automation [58]. Some extraction methods are sufficiently specific and the extracts don’t need further clean-up. One example is the supercritical fluid extraction (SFE) [39, 98, 142], but unfortunately this approach is quite expensive and is thus not expected to gain broad acceptance. Solid-phase microextraction (SPME) has been applied to extract some mycotoxins from solid samples without any further purification [23, 27, 126].
There are screening methods that don’t require any sample preparation, e.g. certain immunoaffinity assays [41], electronic noses [84-87] and IR-based methods [76, 77, 80]. They may have an advantage of circumventing this stage and its associated errors, but are instead prone to their own, which have been covered in detail in the appropriate section of this review. Moreover, the unseparated matrix compounds can greatly affect the results, especially in immunoassay methods [43].
6.4. Analytical method bias
Analytical method, broadly speaking, encompasses also the sampling and sample preparation steps, but in this section the focus will be only on the final analytical determination of mycotoxins in the purified extract.
The particular problems connected with the individual techniques have been addressed in detail previously in the appropriate sections of this review, here we provide only a short overview. Many screening methods, but especially immunoaffinity assays, in particular broadly used ELISA, should be used with caution, as they tend to produce false positives or yield too high values due to crossreactivity and matrix interferences [35]. Interferences of course present the problem also in the more definitive techniques: GC, HPLC. Whenever derivatization of analytes is needed prior to detection, the recovery of the reaction should be closely monitored, and the stability of derivatives might also be a serious problem [39, 138, 211]. With LC-MS or LC-MS/MS methods, ionization suppression in the interface due to visible and “invisible” (i.e. coeluting over a broad range of retention times) matrix interferences is a well-recognized problem, sometimes observed also in GC-MS methods [39, 123, 142, 146, 151, 174, 234]. The proposed solutions include extract dilution [142], matrix-matched calibration [124, 150, 174, 234] and use of internal standards [151], preferably isotopically labeled [142, 146, 159, 176, 180, 186]. In multitoxin methods, more than one internal standard should be added [142] - in fact, the best results are expected when using a corresponding isotopically-labeled IS for each analyte!
All methods for the analysis of mycotoxins should be fully validated by establishing compliance to various criteria: selectivity, linearity, limit of detection, limit of quantification, decision limit, detection capability, intra- and interday precision, recovery, accuracy, robustness etc. [142, 294]. However, this may present additional complications. The first problem is the lack of the calibrants, i.e. sufficiently purified mycotoxin standards. One common problem with the preparation of analytically pure standards is the assessment of their purity [295, 296]. As was shown in the previous sections, different mycotoxins are to a variable extent amenable to various types of detection, while the impurities are completely unpredictable. The search continues for a universal, analyte-independent detector that would have the same response for standard compound and the impurities. Evaporative light-scattering detector has been proposed as such [138], although it doesn’t meet all criteria. Other types of “universal” detectors showing promise are pulsed-discharge photoionization detector and charged aerosol detector (CAD). Another approach is the combined analysis of prepared calibrants by very different methods, e.g. NMR, GC-MS, LC-MS, differential scanning calorimetry etc. [295], or by intercomparison studies in which several experienced laboratories participate [297].
Even with available mycotoxin standards, matrix-matched calibration, which is absolutely necessary [124, 150, 174, 234], may present problems such as interaction between added standard compound and components of the matrix, causing decreasing recoveries [263]. Therefore, the golden standard of validation, certified reference materials (CRMs) should be used in the process of method validation, but not many of them are available at the moment [39, 277, 294, 296, 297]. Interlaboratory studies and comparisons also help to reveal potential methodological problems with mycotoxin analysis in individual laboratories [39, 273, 296, 297].
7. Conclusions
The review presented here covers an enormous subject of various aspects of mycotoxin analysis, which can be readily estimated from the number of references and considering that only the interval from 1998 to present has been covered, but by no means exhaustively. Thus, we hope that we have given an overview of the current status of analytical methods for mycotoxin determination in the broadest sense, and also hinted at the trends that might be promising and take over in the future. Various problems of the mycotoxin analysis have also been tackled. However, this review can’t afford to go into too much depth due to the broadness of the subject; the interested reader is therefore directed to the other excellent reviews that focus more on certain groups of mycotoxins [16, 17, 19, 25, 26, 39, 41, 98, 136-140, 262, 266, 293] or the applied analytical methods [12, 17, 34, 41, 52, 59, 69, 136, 142- 145, 189, 296].
Acknowledgements
This work has been supported by the Slovenian Research Agency (Programme P1-0153). Authors also wish to thank the anonymous reviewers for their helpful suggestions.
This article was originally published in International Journal of Molecular Sciences 2009, 10, 62-115; doi:10.3390/ijms10010062. This is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
References and Notes
1. Richard, J.L. Some major mycotoxins and their mycotoxicoses - An overview. Intern. J. Food Microbiol. 2007, 119, 3–10.
2. Morgavi, D.P.; Riley, R.T. An historical overview of field disease outbreaks known or suspected to be caused by consumption of feeds contaminated with Fusarium toxins. Animal Feed Sci. Technol. 2007, 137, 201–212.
3. Kan, C.A.; Meijer, G.A.L. The risk of contamination of food with toxic substances present in animal feed. Animal Feed Sci. Technol. 2007,133, 84–108.
4. Berg, T. How to establish international limits for mycotoxins in food and feed? Food Control 2003, 14, 219–224.
5. Medina, Á.; Mateo, R.; Valle-Algarra, F.M.; Mateo, E.M.; Jiménez, M. Effect of carbendazim and physicochemical factors on the growth and ochratoxin A production of Aspergillus carbonarius isolated from grapes. Intern. J. Food Microbiol. 2007, 119, 230–235.
6. Orsi, R.B.; Corrêa, B.; Possi, C.R.; Schammass, E.A.; Noueira, J.R.; Dias, S.M.C.; Malozzi, M.A.B. Mycoflora and occurrence of fumonisins in freshly harvested and stored hybrid maize. J. Stored Prod. Res. 2000, 36, 75–87.
7. Wagacha, J.M.; Muthomi, J.W. Mycotoxin problem in Africa: Current status, implications to food safety and health and possible management strategies. Intern. J. Food Microbiol. 2008, 124, 1–12.
8. Marvin, H.J.P.; Kleter, G.A.; Prandini, A.; Dekkers, S.; Bolton, D.J. Early identification systems for emerging foodborne hazards. Food Chem. Toxicol. 2008, DOI: 10.1016/j.fct.2007.12.021.
9. Bullerman, L.B.; Bianchini, A. Stability of mycotoxins during food processing. Intern. J. Food Microbiol. 2007, 119, 140–146.
10. van Egmond, H.P.; Schothorst, R.C.; Jonker, M.A. Regulations relating to mycotoxins in food. Anal. Bioanal. Chem. 2007, 389, 147–157.
11. Shoemaker, R.C.; House, D.E. A time-series study of sick building syndrome: chronic, biotoxinassociated illness from exposure to water-damaged buildings. Neurotoxicol. Teratol. 2005, 27, 29–46.
12. Gutarowska, B.; Piotrowska, M. Methods of mycological analysis in buildings. Build. Environ. 2007, 42, 1843–1850.
13. Tarín, A.; Rosell, M.G.; Guardino, X. Use of high-performance liquid chromatography to assess airborne mycotoxins – Aflatoxins and ochratoxin A. J. Chromatogr. A 2004, 1047, 235–240.
14. Wang, Y.; Chai, T.; Lu, G.; Quan, C.; Duan, H.; Yao, M.; Zucker, B.-A.; Schlenker, G. Simultaneous detection of airborne Aflatoxin, Ochratoxin and Zearalenone in a poultry house by immunoaffinity clean-up and high-performance liquid chromatography. Environ. Res. 2008, 107, 139–144.
15. Shephard, G.S. Determination of mycotoxins in human foods. Chem. Soc. Rev. 2008, 37, 2468–2477.
16. Berthiller, F.; Sulyok, M.; Krska, R.; Schuhmacher, R. Chromatographic methods for the simultaneous determination of mycotoxins and their conjugates in cereals. Intern. J. Food Microbiol. 2007, 119, 33–37.
17. Rundberget, T.; Wilkins, A.L. Determination of Penicillium mycotoxins in foods and feeds using liquid chromatography-mass spectrometry. J. Chromatogr. A 2002, 964, 189–197.
18. Nielsen, K.F.; Sumarah, M.W.; Frisvad, J.C.; Miller, J.D. Production of metabolites from the Penicillium roqueforti complex. J. Agric. Food Chem. 2006, 54, 3756–3763.
19. Xu, B.-J.; Jia, X.-Q.; Gu, L.-J.; Sung, C.-K. Review on the qualitative and quantitative analysis of the mycotoxin citrinin. Food Control 2006, 17, 271–285.
20. Hayashi, Y.; Yoshizawa, T. Analysis of cyclopiazonic acid in corn and rice by a newly developed method. Food Chem. 2005, 93, 215–221.
21. Vrabcheva, T.; Petkova-Bocharova, T.; Grosso, F.; Nikolov, I.; Chernozemsky, I.N.; Castegnaro, M.; Dragacci, S. Analysis of ochratoxin A in foods consumed by inhabitants from an area with Balkan Endemic Nephropathy: A 1 month follow-up study. J. Agric. Food Chem. 2004, 52, 2404–2410.
22. Orem, W.H.; Feder, G.L.; Finkelman, R.B. A possible link between Balkan endemic nephropathy and the leaching of toxic organic compounds from Pliocene lignite by groundwater: Preliminary investigation. Intern. J. Coal Geol. 1999, 40, 237–252.
23. Zambonin, C.G.; Monaci, L.; Aresta, A. Solid-phase microextraction-high performance liquid chromatography and diode array detection for the determination of mycophenolic acid in cheese. Food Chem. 2002, 78, 249–254.
24. Noroozian, E.; Lagerwerf, F.; Lingeman, H.; Brinkman, U.A.Th.; Kerkhoff, M.A.T. Determination of roquefortine C in blue cheese using on-line column-switching liquid chromatography. J. Pharm. Biomed. Anal. 1999, 20, 611–619.
25. Shephard, G.S.; Leggott, N.L. Chromatographic determination of the mycotoxin patulin in fruit and fruit juices. J. Chromatogr. A 2000, 882, 17–22.
26. Uhlig, S.; Torp, M.; Heier, B.T. Beauvericin and enniatins A, A1, B and B1 in Norwegian grain: A survey. Food Chem. 2006, 94, 193–201.
27. Zambonin, C.G.; Monaci, L.; Aresta, A. Determination of cyclopiazonic acid in cheese samples using solid-phase microextraction and high performance liquid chromatography. Food Chem. 2001, 75, 249–254.
28. Lagana, A.; Bacaloni, A.; Castellano, M.; Curini, R.; De Leva, I.; Faberi, A.; Materazzi, S. Sample preparation for determination of macrocyclic lactone mycotoxins in fish tissue, based on on-line matrix solid-phase dispersion and solid-phase extraction cleanup followed by liquid chromatography/tandem mass spectrometry. J. AOAC Intern. 2003, 86, 729–736.
29. Navas, S.A.; Sabino, M.; Rodriguez-Amaya, D.B. Aflatoxin M-1 and ochratoxin A in a human milk bank in the city of Sao Paulo, Brazil. Food Addit. Contam. 2005, 22, 457–462.
30. Gilbert, J.; Brereton, P.; MacDonald, S. Assessment of dietary exposure to ochratoxin A in the UK using a duplicate diet approach and analysis of urine and plasma samples. Food Addit. Contam. 2001, 18, 1088–1093.
31. Mortensen, G.K.; Strobel, B.W.; Hansen, H.C.B. Determination of zearalenone and ochratoxin A in soil. Anal. Bioanal. Chem. 2003, 376, 98–101.
32. Mortensen, G.K.; Strobel, B.W.; Hansen, H.C.B. Degradation of zearalenone and ochratoxin A in three Danish agricultural soils. Chemosphere 2006, 62, 1673–1680.
33. Lagana, A.; Fago, G.; Marino, A.; Santarelli, D. Development of an analytical system for the simultaneous determination of anabolic macrocyclic lactones in aquatic environmental samples. Rapid Comm. Mass Spectrom. 2001, 15, 304–310.
34. Prieto-Simón, B.; Noguer, T.; Campàs, M. Emerging biotools for assessment of mycotoxins in the past decade. TrAC 2007, 26, 689–702.
35. Shephard, G.S.; van der Westhuizen, L.; Gatyeni, P.M.; Katerere, D.R.; Marasas, W.F.O. Do fumonisin mycotoxins occur in wheat? J. Agric. Food Chem. 2005, 53, 9293–9296.
36. Erbs, M.; Hartmann, N.; Bucheli, T.D. Determination of the cross-reactivities for alpha-zearalenol, beta-zearalenol, zearalanone, alpha-zearalanol, and beta-zearalanol on three commercial immunoaffinity columns targeting zearalenone. J. AOAC Intern. 2007, 90, 1197–1202.
37. Yu, F.-Y.; Chi, T.-F.; Liu, B.-H.; Su, C.-C. Development of a sensitive enzyme-linked immunosorbent assay for the determination of ochratoxin A. J. Agric. Food Chem. 2005, 53, 6947–6953.
38. Lauer, B.; Ottleben, I.; Jacobsen, H.-J.; Reinard, T. Production of a single-chain variable fragment antibody against fumonisin B1. J. Agric. Food Chem. 2005, 53, 899–904.
39. Krska, R.; Welzig, E.; Boudra, H. Analysis of Fusarium toxins in feed. Animal Feed Sci. Technol. 2007, 137, 241–264.
40. Zheng, M.Z.; Richard, J.L.; Binder, J. A review of rapid methods for the analysis of mycotoxins. Mycopathologia 2006, 161, 261–273.
41. Schneider, E.; Curtui, V.; Seidler, C.; Dietrich, R.; Usleber, E.; Märtlbauer, E. Rapid methods for deoxynivalenol and other trichothecenes. Toxicol. Lett. 2004, 153, 113–121.
42. Badea, M.; Micheli, L.; Messia, M.C.; Candigliota, T.; Marconi, E.; Mottram, T.; Velasco-Garcia, M.; Moscone, D.; Palleschi, G. Aflatoxin M1 determination in raw milk using a flow-injection immunoassay system. Anal. Chim. Acta 2004, 520, 141–148.
43. Piermarini, S.; Volpe, G.; Micheli, L.; Moscone, D.; Palleschi, G. An ELIME-array for detection of aflatoxin B1 in corn samples. Food Control 2009, 20, 371–375.
44. Pal, A.; Dhar, T.K. An analytical device for on-site immunoassay. Demonstration of its applicability in semiquantitative detection of aflatoxin B-1 in a batch of samples with ultrahigh sensitivity. Anal. Chem. 2004, 76, 98–104.
45. Urraca, J.L.; Benito-Peña, E.; Pérez-Conde, C.; Moreno-Bondi, M.C.; Pestka, J.J. Analysis of zearalenone in cereal and swine feed samples using an automated flow-through immunosensor. J. Agric. Food Chem. 2005, 53, 3338–3344.
46. de Champdoré, M.; Bazzicalupo, P.; De Napoli, L.; Montesarchio, D.; Di Fabio, G.; Cocozza, I.; Parracino, A.; Rossi, M.; D'Auria, S. A new competitive fluorescence assay for the detection of patulin toxin. Anal. Chem. 2007, 79, 751–757.
47. Nasir, M.S.; Jolley, M.E. Development of a fluorescence polarization assay for the determination of aflatoxins in grains. J. Agric. Food Chem. 2002, 50, 3116–3121.
48. Mullett, W.; Lai, E.P.C.; Yeung, J.M. Immunoassay of fumonisins by a surface plasmon resonance biosensor. Anal. Biochem. 1998, 258, 161–167.
49. van der Gaag, B.; Spath, S.; Dietrich, H.; Stigter, E.; Boonzaaijer, G.; van Osenbruggen, T.; Koopal, K. Biosensors and multiple mycotoxin analysis. Food Control 2003, 14, 251–254.
50. Piermarini, S.; Micheli, L.; Ammida, N.H.S.; Palleschi, G.; Moscone, D. Electrochemical immunosensor array using a 96-well screen-printed microplate for aflatoxin B-1 detection. Biosens. Bioelectr. 2007, 6, 1434–1440.
51. Ho, J.A.; Durst, R.A. Detection of fumonisin B1: Comparison of flow-injection liposome immunoanalysis with high-performance liquid chromatography. Anal. Biochem. 2003, 6, 7–13.
52. Goryacheva, I.Y.; De Saeger, S.; Eremin, S.A.; Van Peteghem, C. Immunochemical methods for rapid mycotoxin detection: Evolution from single to multiple analyte screening: A review. Food Addit. Contam. 2007, 6, 1169–1183.
53. Adányi, N.; Levkovets, I.A.; Rodriguez-Gil, S.; Ronald, A.; Váradi, M.; Szendro, I. Development of immunosensor based on OWLS technique for determining Aflatoxin B1 and Ochratoxin A. Biosens. Bioelectr. 2007, 6, 797–802.
54. Saha, D.; Acharya, D.; Roy, D.; Shrestha, D.; Dhar, T.K. Simultaneous enzyme immunoassay for the screening of aflatoxin B-1 and ochratoxin A in chili samples. Anal. Chim. Acta 2007, 584, 343–349.
55. Köller, G.; Wichmann, G.; Rolle-Kampczyk, U.; Popp, P.; Herbarth, O. Comparison of ELISA and capillary electrophoresis with laser-induced fluorescence detection in the analysis of Ochratoxin A in low volumes of human blood serum. J. Chromatogr. B 2006, 840, 94–98.
56. Papadopoulou-Bouraoui, A.; Vrabcheva, T.; Valzacchi, S.; Stroka, J.; Anklam, E. Screening survey of deoxynivalenol in beer from the European market by an enzyme-linked immunosorbent assay. Food Addit. Contam. 2004, 21, 607–617.
57. Goryacheva, I.Y.; De Saeger, S.; Nesterenko, I.S.; Eremin, S.A.; Van Peteghem, C. Rapid all-inone three-step immunoassay for non-instrumental detection of ochratoxin A in high-coloured herbs and spices. Talanta 2007, 72, 1230–1234.
58. Rodriguez-Mozaz, S.; Lopez de Alda, M.J.; Barceló, D. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography-mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J. Chromatogr. A 2007, 1152, 97–115.
59. Logrieco, A.; Arrigan, D.W.M.; Brengel-Pesce, K.; Siciliano, P.; Tothill, I. DNA arrays, electronic noses and tongues, biosensors and receptors for rapid detection of toxigenic fungi and mycotoxins: A review. Food Addit. Contam. 2005, 22, 335–344.
60. Schnerr, H.; Vogel, R.F.; Niessen, L. A biosensor-based immunoassay for rapid screening of deoxynivalenol contamination in wheat. Food Agric. Immunol. 2002, 14, 313–321.
61. Ngundi, M.M.; Shriver-Lake, L.C.; Moore, M.H.; Lassman, M.E.; Ligler, F.S.; Taitt, C.R. Array biosensor for detection of ochratoxin A in cereals and beverages. Anal. Chem. 2005, 77, 148–154.
62. Ammida, N.H.S.; Micheli, L.; Piermarini, S.; Moscone, D.; Palleschi, G. Detection of aflatoxin B- 1 in barley: Comparative study of immunosensor and HPLC. Anal. Lett. 2006, 39, 1559–1572.
63. Micheli, L.; Grecco, R.; Badea, M.; Moscone, D.; Palleschi, G. An electrochemical immunosensor for aflatoxin M1 determination in milk using screen-printed electrodes. Biosens. Bioelectr. 2005, 21, 588–596.
64. Owino, J.H.O.; Ignaszak, A.; Al-Ahmed, A.; Baker, P.G.L.; Alemu, H.; Ngila, J.C.; Iwuoha, E.I. Modelling of the impedimetric responses of an aflatoxin B-1 immunosensor prepared on an electrosynthetic polyaniline platform. Anal. Bioanal. Chem. 2007, 388, 1069–1074.
65. Baggiani, C.; Anfossi, L.; Giovannoli, C. Molecular imprinted polymers as synthetic receptors for the analysis of myco- and phyco-toxins. Analyst 2008, 133, 719–730.
66. Urraca, J.L.; Marazuela, M.D.; Merino, E.R.; Orellana, G.; Moreno-Bondi, M.C. Molecularly imprinted polymers with a streamlined mimic for zearalenone analysis. J. Chromatogr. A 2006, 1116, 127–134.
67. Navarro-Villoslada, F.; Urraca, J.L.; Moreno-Bondi, M.C.; Orellana, G. Zearalenone sensing with molecularly imprinted polymers and tailored fluorescent probes. Sens. Actuat. B 2007, 121, 67–73.
68. Maier, N.M.; Buttinger, G.; Welhartizki, S.; Gavioli, E.; Lindner, W. Molecularly imprinted polymer-assisted sample clean-up of ochratoxin A from red wine: merits and limitations. J. Chromatogr. B 2004, 804, 103–111.
69. Lin, L.; Zhang, J.; Wang, P.; Wang, Y.; Chen, J. Thin-layer chromatography of mycotoxins and comparison with other chromatographic methods. J. Chromatogr. A 1998, 815, 3–20.
70. Peña, R.; Alcaraz, M.C.; Arce, L.; Ríos, A.; Valcárcel, M. Screening of aflatoxins in feed samples using a flow system coupled to capillary electrophoresis. J. Chromatogr. A 2002, 967, 303–314.
71. Zougagh, M.; Ríos, A. Supercritical fluid extraction of macrocyclic lactone mycotoxins in maize flour samples for rapid amperometric screening and alternative liquid chromatographic method for confirmation. J. Chromatogr. A 2008, 1177, 50–57.
72. Malone, B.R.; Humphrey, C.W.; Romer, T.R.; Richard, J.L. One-step solid-phase extraction cleanup and fluorometric analysis of deoxynivalenol in grains. J. AOAC Intern. 1998, 81, 448–452.
73. Arduini, F.; Errico, I.; Amine, A.; Micheli, L.; Palleschi, G.; Moscone, D. Enzymatic spectrophotometric method for aflatoxin B detection based on acetylcholinesterase inhibition. Anal. Chem. 2007, 79, 3409–3415.
74. Engler, K.H.; Coker, R.; Evans, I.H. A novel colorimetric yeast bioassay for detecting trichothecene mycotoxins. J. Microbiol. Meth. 1999, 35, 207–218.
75. Abramovic, B.; Jajic, I.; Abramovic, B.; Cosic, J.; Juric, V. Detection of deoxynivalenol in wheat by Fourier transform infrared spectroscopy. Acta Chim. Slov. 2007, 54, 859–867.
76. Kos, G.; Lohninger, H.; Krska, R. Fourier transform mid-infrared spectroscopy with attenuated total reflection (FT-IR/ATR) as a tool for the detection of Fusarium fungi on maize. Vibr. Spectrosc. 2002, 29, 115–119. 7
7. Kos, G.; Lohninger, H.; Mizaikoff, B.; Krska, R. Optimisation of a sample preparation procedure for the screening of fungal infection and assessment of deoxynivalenol content in maize using mid-infrared attenuated total reflection spectroscopy. Food Addit. Contam. 2007, 24, 721–729.
78. Galvis-Sánchez, A.C.; Barros, A.; Delgadillo, I. FTIR-ATR infrared spectroscopy for the detection of ochratoxin A in dried vine fruit. Food Addit. Contam. 2007, 24, 1299–1305.
79. Galvis-Sánchez, A.C.; Barros, A.S.; Delgadillo, I. Method for analysis dried vine fruits contaminated with ochratoxin A. Anal. Chim. Acta 2008, 617, 59–63.
80. Pettersson, H.; Åberg, L. Near infrared spectroscopy for determination of mycotoxins in cereals. Food Control 2003, 14, 229–232.
81. Hernández-Hierro, J.M.; García-Villanova, R.J.; González-Martín, I. Potential of near infrared spectroscopy for the analysis of mycotoxins applied to naturally contaminated red paprika found in the Spanish market. Anal. Chim. Acta 2008, 622, 189–194.
82. Demyttenaere, J.C.R.; Moriña, R-M.; Sandra, P. Monitoring and fast detection of mycotoxinproducing fungi based on headspace solid-phase microextraction and headspace sorptive extraction of the volatile metabolites. J. Chromatogr. A 2003, 985, 127–135.
83. Demyttenaere, J.C.; Moriña, R.M.; De Kimpe, N.; Sandra, P. Use of headspace solid-phase microextraction and headspace sorptive extraction for the detection of the volatile metabolites produced by toxigenic Fusarium species. J. Chromatogr. A 2004, 1027, 147–154.
84. Paolesse, R.; Alimelli, A.; Martinelli, E.; Di Natale, C.; D'Amico, A.; D'Egidio, M.G.; Aureli, G.; Ricelli, A.; Fanelli, C. Detection of fungal contamination of cereal grain samples by an electronic nose. Sens. Act. B 2006, 119, 425–430.
85. Presicce, D.S.; Forleo, A.; Taurino, A.M.; Zuppa, M.; Siciliano, P.; Laddomada, B.; Logrieco, A.; Visconti, A. Response evaluation of an E-nose towards contaminated wheat by Fusarium poae fungi. Sens. Act. B 2006, 118, 433–438.
86. Karlshøj, K.; Nielsen, P.V.; Larsen, T.O. Prediction of Penicillium expansum spoilage and patulin concentration in apples used for apple juice production by electronic nose analysis. J. Agric. Food Chem. 2007, 55, 4289–4298.
87. Olsson, J.; Börjesson, T.; Lundstedt, T.; Schnürer, J. Detection and quantification of ochratoxin A and deoxynivalenol in barley grains by GC-MS and electronic nose. Intern. J. Food Microbiol. 2002, 72, 203–214.
88. Schnurer, J.; Olsson, J.; Borjesson, T. Fungal volatiles as indicators of food and feeds spoilage. Fungal. Genet. Biol. 1999, 27, 209–217.
89. Janardhana G.R.; Raveesha K.A.; Shetty H.S. Mycotoxin contamination of maize grains grown in Karnataka (India). Food Chem. Toxicol. 1999, 37, 863–868.
90. Park J.W.; Shon D.H.; Kim Y.B. Application of an enzyme-linked immunosorbent assay for detecting mold contamination in agricultural commodities and comparison with conventional assays. Food Agric. Immunol. 2003, 15, 159–166
. 91. Hartmann, N.; Erbs, M.; Wettstein, F.E.; Hoerger, C.C.; Schwarzenbach, R.P.; Bucheli, T.D. Quantification of zearalenone in various solid agroenvironmental samples using D-6-Zearalenone as the internal standard. J. Agric. Food Chem. 2008, 56, 2926–2932.
92. Blaney, B.J.; Maryam, R.; Murray, S.A.; Ryley, M.J. Alkaloids of the sorghum ergot pathogen (Claviceps africana): Assay methods for grain and feed and variation between sclerotia/sphacelia. Aust. J. Agr. Res. 2003, 54, 167–175.
93. Urraca, J.L.; Marazuela, M.D.; Moreno-Bondi, M.C. Analysis for zearalenone and alphazearalenol in cereals and swine feed using accelerated solvent extraction and liquid chromatography with fluorescence detection. Anal. Chim. Acta 2004, 524, 175–183.
94. Gonzalez, L.; Juan, C.; Soriano, J.M.; Molto, J.C.; Manes, J. Occurrence and daily intake of ochratoxin A of organic and non-organic rice and rice products. Intern. J. Food Microbiol. 2006, 107, 223–227.
95. Sheibani, A.; Ghaziaskar H.S. Pressurized fluid extraction for quantitative recovery of aflatoxins B1 and B2 from pistachio. Food Control 2009, 20, 124–128.
96. Royer D.; Humpf H.U.; Guy P.A. Quantitative analysis of Fusarium mycotoxins in maize using accelerated solvent extraction before liquid chromatography atmospheric pressure chemical ionization tandem mass spectrometry. Food Addit. Contam. 2004, 21, 678–692.
97. Pallaroni, L.; von Holst, C.; Eskilsson, C.S.; Bjorklund, E. Microwave-assisted extraction of zearalenone from wheat and corn. Anal. Bioanal. Chem. 2002, 374, 161–166.
98. Krska, R. Performance of modern sample preparation techniques in the analysis of Fusarium mycotoxins in cereals. J. Chromatogr. A 1998, 815, 49–57.
99. Ahmed, N.E.; Farag, M.M.; Soliman, K.M.; Abdel-Samed, A.K.M.; Naguib, Kh.M. Evaluation of methods used to determine ochratoxin a in coffee beans. J. Agric. Food Chem. 2007, 55, 9576–9580.
100.Urraca J.L.; Carbajo M.C.; Torralvo M.J.; Gonzalez-Vazquez J.; Orellana G.; Moreno-Bondi M.C. Effect of the template and functional monomer on the textural properties of molecularly imprinted polymers. Biosens. Bioelectron. 2008, 24, 155–161.
101.Reinsch, M.; Topfer, A.; Lehmann, A.; Nehls, I. Determination of ochratoxin A in wine by liquid chromatography tandem mass spectrometry after combined anion-exchange/reversed-phase cleanup. Anal. Bioanal. Chem. 2005, 381, 1592–1595.
102.Launay, F.M.; Young, P.B.; Sterk, S.S.; Blokland, M.H.; Kennedy, D.G. Confirmatory assay for zeranol, taleranol and the Fusarium spp. toxins in bovine urine using liquid chromatographytandem mass spectrometry. Food Addit. Contam. 2004, 21, 52–62.
103.Garcia-Villanova, R.; Cordón, C.; González Paramás, A.M.; Aparicio, P.; Garcia Rosales, M.E. Simultaneous immunoaffinity column cleanup and HPLC analysis of aflatoxins and ochratoxin A in Spanish bee pollen. J. Agric. Food Chem. 2004, 52, 7235–7239.
104.Kruger, S.C.; Kohn, B.; Ramsey, C.S.; Prioli R. Rapid immunoaffinity-based method for determination of zearalenone in corn by fluorometry and liquid chromatography. J. AOAC Intern. 1999, 82, 1364–1368.
105.Calleri, E.; Marrubini, G.; Brusotti, G.; Massolini, G.; Caccialanza, G. Development and integration of an immunoaffinity monolithic disk for the on-line solid-phase extraction and HPLC determination with fluorescence detection of aflatoxin B1 in aqueous solutions. J. Pharm. Biomed. Anal. 2007, 44, 396–403.
106.Trebstein, A.; Seefelder, W.; Lauber, U.; Humpf, H.-U. Determination of T-2 and HT-2 toxins in cereals including oats after immunoaffinity cleanup by liquid chromatography and fluorescence detection. J. Agric. Food Chem. 2008, 56, 4968–4975.
107.Brenn-Struckhofova Z.; Cichna-Markl M.; Bohm C.; Razzazi-Fazeli E. Selective sample cleanup by reusable sol-gel immunoaffinity columns for determination of deoxynivalenol in food and feed samples. Anal. Chem. 2007, 79, 710–717.
108.Sharma, M.; Márquez, C. Determination of aflatoxins in domestic pet foods (dog and cat) using immunoaffinity column and HPLC. Anim. Feed Sci. Technol. 2001, 93, 109–114.
109.Songsermsakul, P.; Sontag, G.; Cichna-Markl, M.; Zentek, J.; Razzazi-Fazeli, E. Determination of zearalenone and its metabolites in urine, plasma and faeces of horses by HPLC-APCI-MS. J. Chromatogr. B 2006, 843, 252–261.
110.Giraudi, G.; Anfossi, L.; Baggiani, C.; Giovannoli, C.; Tozzi, C. Solid-phase extraction of ochratoxin A from wine based on a binding hexapeptide prepared by combinatorial synthesis. J. Chromatogr. A 2007, 1175, 174–180.
111.Chan, D.; MacDonald, S.J.; Boughtflower, V.; Brereton, P. Simultaneous determination of aflatoxins and ochratoxin A in food using a fully automated immunoaffinity column clean-up and liquid chromatography-fluorescence detection. J. Chromatogr. A 2004, 1059, 13–16.
112.Sáez, J.M.; Medina, Á.; Gimeno-Adelantado, J.V.; Mateo, R.; Jiménez, M. Comparison of different sample treatments for the analysis of ochratoxin A in must, wine and beer by liquid chromatography. J. Chromatogr. A 2004, 1029, 125–133.
113.Baggiani, C.; Anfossi, L.; Giovannoli, C. Solid phase extraction of food contaminants using molecular imprinted polymers. Anal. Chim. Acta 2007, 591, 29–39.
114.Pascale, M.; De Girolamo, A.; Visconti, A.; Magan, N.; Chianella, I.; Piletska, E.V.; Piletsky, S.A. Use of itaconic acid-based polymers for solid-phase extraction of deoxynivalenol and application to pasta analysis. Anal. Chim. Acta 2008, 609, 131–138.
115.Appell, M.; Kendra, D.F.; Kim, E.K.; Maragos, C.M. Synthesis and evaluation of molecularly imprinted polymers as sorbents of moniliformin. Food Addit. Contam. 2007, 24, 43–52.
116.Visconti, A.; Pascale, M. Determination of zearalenone in corn by means of immunoaffinity clean-up and high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 1998, 815, 133–140.
117.Tomaševic-Canovic, M.; Dakovic, A.; Rottinghaus G.; Matijaševic, S.; Ðuricic, M. Surfactant modified zeolites - new efficient adsorbents for mycotoxins. Micropor. Mesopor. Mat. 2003, 61, 173–180.
118.Kralj Cigic, I.; Strlic, M.; Schreiber, A.; Kocjancic, M.; Pihlar, B. Ochratoxin A in wine: Its determination and photostability. Anal. Lett. 2006, 39, 1475–1488.
119.Blesa, J.; Soriano, J.M.; Moltó, J.C.; Marín, R.; Mañes, J. Determination of aflatoxins in peanuts by matrix solid-phase dispersion and liquid chromatography. J. Chromatogr. A 2003, 1011, 49–54.
120.Gentili, A.; Caretti, F.; D'Ascenzo, G.; Rocca L.M.; Marchese S.; Materazzi S.; Perret D. Simultaneous determination of trichothecenes A, B, and D in maize food products by LC-MSMS. Chromatographia 2007, 66, 669–676.
121.Sulyok, M.; Krska, R.; Schuhmacher, R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semiquantitative screening of moldy food samples. Anal. Bioanal. Chem. 2007, 389, 1505–1523.
122.Xavier, J.J.M.; Scussel, V.M. Development of an LC-MS/MS method for the determination of aflatoxins B-1, B-2, G(1), and G(2) in Brazil nut. Intern. J. Envir. Anal. Chem. 2008, 88, 425–433.
123.Spanjer, M.C.; Rensen, P.M.; Scholten, J.M. LC-MS/MS multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit. Contam. 2008, 25, 472–489.
124.Sulyok, M.; Krska, R.; Schuhmacher, R. Application of a liquid chromatography-tandem mass spectrometric method to multi-mycotoxin determination in raw cereals and evaluation of matrix effects. Food Addit. Contam. 2007, 24, 1184–1195.
125.Zollner P.; Leitner A.; Lubda D.; Cabrera K.; Lindner W. Application of a chromolith SpeedROD RP-18e HPLC column: Determination of ochratoxin A in different wines by high-performance liquid chromatography-tandem mass spectrometry. Chromatographia 2000, 52, 818–820.
126.Vatinno, R.; Aresta, A.; Zambonin, C.G.; Palmisano, F. Determination of Ochratoxin A in green coffee beans by solid-phase microextraction and liquid chromatography with fluorescence detection. J. Chromatogr. A 2008, 1187, 145–150.
127.Vatinno, R.; Vuckovic, D.; Zambonin, C.G.; Pawliszyn, J. Automated high-throughput method using solid-phase microextraction-liquid chromatography-tandem mass spectrometry for the determination of ochratoxin A in human urine. J. Chromatogr. A 2008, 1201, 215–221.
128.Boehm, C.; Cichna-Markl, M.; Brenn-Struckhofova, Z.; Razzazi-Fazeli, E. Development of a selective sample clean-up method based on immuno-ultrafiltration for the determination of deoxynivalenol in maize. J. Chromatogr. A 2008, 1202, 111–117.
129.Sheu, F.; Shyu, Y.T. Analysis of patulin in apple juice by diphasic dialysis extraction with in situ acylation and mass spectrometric determination. J. Agric. Food Chem. 1999, 47, 2711–2714.
130.García-Fonseca, S.; Ballesteros-Gómez, A.; Rubio, S.; Pérez-Bendito, D. Coacervative extraction of Ochratoxin A in wines prior to liquid chromatography/fluorescence determination. Anal. Chim. Acta 2008, 617, 3–10.
131.Almeda, S.; Arce, L.; Valcarcel, M. Combined use of supported liquid membrane and solid-phase extraction to enhance selectivity and sensitivity in capillary electrophoresis for the determination of ochratoxin A in wine. Electrophoresis 2008, 29, 1573–1581.
132.Russo, M.V.; Veschetti, E.; Cinelli, G.; Avino, P. Short capillary traps in GC-GC tandem systems for direct analysis of T2 mycotoxin in aqueous samples. Chromatographia 2007, 66, 237–242.
133.Medina, A.; Valle-Algarra, F.M.; Gimeno-Adelantado, J.V.; Mateo, R.; Mateo, F.; Jimenez, M. New method for determination of ochratoxin A in beer using zinc acetate and solid-phase extraction silica cartridges. J. Chromatogr. A 2006, 1121, 178–183.
134.Medina, Á.; Jiménez, M.; Gimeno-Adelantado, J.V.; Valle-Algarra, F.M.; Mateo, R. Determination of ochratoxin A in beer marketed in Spain by liquid chromatography with fluorescence detection using lead hydroxyacetate as a clean-up agent. J. Chromatogr. A 2005, 1083, 7–13.
135.Onji, Y.; Aoki, Y.; Tani, N.; Umebayashi, K.; Kitada, Y.; Dohi, Y. Direct analysis of several Fusarium mycotoxins in cereals by capillary gas chromatography mass spectrometry. J. Chromatogr. A 1998, 815, 59–65.
136.Langseth, W.; Rundberget, T. Instrumental methods for determination of nonmacrocyclic trichothecenes in cereals, foodstuffs and cultures. J. Chromatogr. A 1998, 815, 103–121.
137.Valenta, H. Chromatographic methods for the determination of ochratoxin A in animal and human tissues and fluids. J. Chromatogr. A 1998, 815, 75–92.
138.Wilkes, J.G.; Sutherland, J.B. Sample preparation and high-resolution separation of mycotoxins possessing carboxyl groups. J. Chromatogr. B 1998, 717, 135–156. 139.Shephard, G.S. Chromatographic determination of the fumonisin mycotoxins. J. Chromatogr. A 1998, 815, 31–39.
140.Krska, R.; Josephs, R. The state-of-the-art in the analysis of estrogenic mycotoxins in cereals. Fresenius J. Anal. Chem. 2001, 369, 469–476.
141.Sforza, S.; Dall'Asta, C.; Marchelli, R. Recent advances in mycotoxin determination in food and feed by hyphenated chromatographic techniques/mass spectrometry. Mass Spectrom. Rev. 2006, 25, 54–76.
142.Zöllner, P.; Mayer-Helm, B. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A 2006, 1136, 123–169.
143.Songsermsakul, P.; Razzazi-Fazeli, E. A review of recent trends in applications of liquid chromatography-mass spectrometry for determination of mycotoxins. J. Liq. Chrom. Rel. Techn. 2008, 31, 1641–1686.
144.Krska, R.; Molinelli, A. Mycotoxin analysis: State-of-the-art and future trends. Anal. Bioanal. Chem. 2007, 387, 145–148.
145.Krska, R.; Schubert-Ullrich, P.; Molinelli, A.; Sulyok, M.; Macdonald, S.; Crews, C. Mycotoxin analysis: An update. Food Addit. Contam. 2008, 25, 152–163.
146.Razzazi-Fazeli, E.; Rabus, B.; Cecon, B.; Böhm, J. Simultaneous quantification of Atrichothecene mycotoxins in grains using liquid chromatography atmospheric pressure chemical ionisation mass spectrometry. J. Chromatogr. A 2002, 968, 129–142.
147.Laganà, A.; Curini, R.; D'Ascenzo, G.; De Leva, I.; Faberi, A.; Pastorini, E. Liquid chromatography/tandem mass spectrometry for the identification and determination of trichothecenes in maize. Rapid Comm. Mass Spectrom. 2003, 17, 1037–1043.
148.Tuomi, T.; Saarinen, L.; Reijula, K. Detection of polar and macrocyclic trichothecene mycotoxins from indoor environments. Analyst 1998, 123, 1835–1841.
149.Razzazi-Fazeli, E.; Böhm, J.; Jarukamjorn, K.; Zentek, J. Simultaneous determination of major Btrichothecenes and the de-epoxy-metabolite of deoxynivalenol in pig urine and maize using highperformance liquid chromatography-mass spectrometry. J. Chromatogr. B 2003, 796, 21–33.
150.Berthiller, F.; Schuhmacher, R.; Buttinger, G.; Krska, R. Rapid simultaneous determination of major type A- and B-trichothecenes as well as zearalenone in maize by high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2005, 1062, 209–216.
151.Klötzel, M.; Gutsche, B, Lauber, U.; Humpf, H.-U. Determination of 12 type A and B trichothecenes in cereals by liquid chromatography-electrospray ionization tandem mass spectrometry. J. Agric. Food Chem. 2005, 53, 8904–8910.
152.Rosenberg, E.; Krska, R.; Wissiack, R.; Kmetov, V.; Josephs, R.; Razzazi, E.; Grasserbauer, M. High-performance liquid chromatography atmospheric-pressure chemical ionization mass spectrometry as a new tool for the determination of the mycotoxin zearalenone in food and feed. J. Chromatogr. A 1998, 819, 277–288.
153.Maragou, N.C.; Rosenberg, E.; Thomaidis, N.S.; Koupparis, M.A. Direct determination of the estrogenic compounds 8-prenylnaringenin, zearalenone, alpha- and beta-zearalenol in beer by liquid chromatography-mass spectrometry. J. Chromatogr. A 2008, 1202, 47–57.
154.Zöllner, P.; Jodlbauer, J.; Lindner, W. Determination of zearalenone in grains by highperformance liquid chromatography-tandem mass spectrometry after solid-phase extraction with RP-18 columns or immunoaffinity columns. J. Chromatogr. A 1999, 858, 167–174.
155.Zöllner, P.; Jodlbauer, J.; Kleinova, M.; Kahlbacher, H.; Kuhn, T.; Hochsteiner, W.; Lindner, W. Concentration levels of zearalenone and its metabolites in urine, muscle tissue, and liver samples of pigs fed with mycotoxin-contaminated oats. J. Agric. Food Chem. 2002, 50, 2494–2501.
156.Hartmann, N.; Erbs, M.; Wettstein, F.E.; Schwarzenbach, R.P.; Bucheli, T.D. Quantification of estrogenic mycotoxins at the ng/L level in aqueous environmental samples using deuterated internal standards. J. Chromatogr. A 2007, 1138, 132–140.
157.Schneweis, I.; Meyer, K.; Engelhardt, G.; Bauer, J. Occurrence of zearalenone-4-beta-Dglucopyranoside in wheat. J. Agric. Food Chem. 2002, 50, 1736–1738.
158.Berthiller, F.; Werner, U.; Sulyok, M.; Krska, R.; Hauser, M.T.; Schuhmacher, R. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) determination of phase II metabolites of the mycotoxin zearalenone in the model plant Arabidopsis thaliana. Food Addit. Contam. 2006, 23, 1194–1200.
159.Cramer, B.; Bretz, M.; Humpf, H.-U. Stable isotope dilution analysis of the Fusarium mycotoxin zearalenone. J. Agric. Food Chem. 2007, 55, 8353–8358.
160.van Bennekom, E.O.; Brouwer, L.; Laurant, E.H.M.; Hooijerink, H.; Nielen, M.W.F. Confirmatory analysis method for zeranol, its metabolites and related mycotoxins in urine by liquid chromatography-negative ion electrospray tandem mass spectrometry. Anal. Chim. Acta 2002, 473, 151–160.
161.Schmidt, K.; Stachel, C.; Gowik, P. Development and in-house validation of an LC-MS/MS method for the determination of stilbenes and resorcylic acid lactones in bovine urine. Anal. Bioanal. Chem. 2008, 391, 1199–1210.
162.Senyuva, H.Z.; Ozcan, S.; Cimen, D.; Gilbert, J. Determination of fumonisins B-1 and b2 in corn by liquid chromatography/mass spectrometry with immunoaffinity column cleanup: Singlelaboratory method validation. J. AOAC Intern. 2008, 91, 598–606.
163.Seefelder, W.; Gossmann, M.; Humpf, H.-U. Analysis of fumonisin B-1 in Fusarium proliferatum-infected asparagus spears and garlic bulbs from Germany by liquid chromatography - Electrospray ionization mass spectrometry. J. Agric. Food Chem. 2002, 50, 2778–2781.
164.Faberi, A.; Foglia, P.; Pastorini, E.; Samperi, R.; Laganà, A. Determination of type B fumonisin mycotoxins in maize and maize-based products by liquid chromatography/tandem mass spectrometry using a QqQ(linear ion trap) mass spectrometer. Rapid Comm. Mass Spectrom. 2005, 19, 275–282.
165.Paepens, C.; De Saeger, S.; Sibanda, L.; Barna-Vetró, I.; Anselme, M.; Larondelle, Y.; Van Peteghem, C. Evaluation of fumonisin contamination in cornflakes on the Belgian market by "flow-through" assay screening and LC-MS/MS analyses. J. Agric. Food Chem. 2005, 53, 7337–7343.
166.Jestoi, M.; Rokka, M.; Rizzo, A.; Peltonen, K.; Aurasaari, S. Determination of Fusariummycotoxins beauvericin and enniatins with liquid chromatography-tandem mass spectrometry (LC-MS/MS). J. Liq. Chrom. Rel. Techn. 2005, 28, 369–381.
167.Sewram, V.; Nieuwoudt, T.W.; Marasas, W.F.O.; Shephard, G.S.; Ritieni, A. Determination of the mycotoxin moniliformin in cultures of Fusarium subglutinans and in naturally contaminated maize by high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 1999, 848, 185–191.
168.Cavaliere, C.; Foglia, P.; Pastorini, E.; Samperi, R.; Laganà, A. Development of a multiresidue method for analysis of major Fusarium mycotoxins in corn meal using liquid chromatography/tandem mass spectrometry. Rapid Comm. Mass Spectrom. 2005, 19, 2085–2093.
169.Cavaliere, C.; Foglia, P.; Guarino, C.; Motto, M.; Nazzari, M.; Samperi, R.; Laganà, A.; Berardo, N. Mycotoxins produced by Fusarium genus in maize: Determination by screening and confirmatory methods based on liquid chromatography tandem mass spectrometry. Food Chem. 2007, 105, 700–710.
170.Engelhart, S.; Loock, A.; Skutlarek, D.; Sagunski, H.; Lommel, A.; Farber, H.; Exner, M. Occurrence of toxigenic Aspergillus versicolor isolates and sterigmatocystin in carpet dust from damp indoor environments. Appl. Environ. Microb. 2002, 68, 3886–3890.
171.Bloom, E.; Bal, K.; Nyman, E.; Must, A.; Larsson, L. Mass spectrometry-based strategy for direct detection and quantification of some mycotoxins produced by Stachybotrys and Aspergillus spp. in indoor environments. Appl. Environ. Microb. 2007, 73, 4211–4217.
172.Tuomi, T.; Johnsson, T.; Hintikka, E.L.; Reijula, K. Detection of aflatoxins (G(1-2), B1-2), sterigmatocystin, citrinine and ochratoxin A in samples contaminated by microbes. Analyst 2001, 126, 1545–1550.
173.Ventura, M.; Guillen, D.; Anaya, I.; Broto-Puig, F.; Lliberia, J.L.; Agut, M.; Comellas, L. Ultraperformance liquid chromatography/tandem mass spectrometry for the simultaneous analysis of aflatoxins B1, G1, B2, G2 and ochratoxin A in beer. Rapid Comm. Mass Spectrom. 2006, 20, 3199–3204.
174.Cervino, C.; Asam, S.; Knopp, D.; Rychlik, M.; Niessner, R. Use of isotope-labeled Aflatoxins for LC-MS/MS stable isotope dilution analysis of foods. J. Agric. Food Chem. 2008, 56, 1873–1879.
175.Becker, M.; Degelmann, P.; Herderich, M.; Schreier, P.; Humpf, H.-U. Column liquid chromatography electrospray ionisation tandem mass spectrometry for the analysis of ochratoxin. J. Chromatogr. A 1998, 818, 260–264.
176.Lindenmeier, M.; Schieberle, P.; Rychlik, M. Quantification of ochratoxin A in foods by a stable isotope dilution assay using high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2004, 1023, 57–66.
177.Chung, S.W.C.; Kwong, K.P. Determination of ochratoxin a at parts-per-trillion levels in cereal products by immunoaffinity column cleanup and high-performance liquid chromatography/mass spectrometry. J. AOAC Intern. 2007, 90, 773–777.
178.Lau, B.P.-Y.; Scott, P.M.; Lewis, D.A.; Kanhere, S.R.; Cléroux, C.; Roscoe, V.A. Liquid chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry of the Altemaria mycotoxins alternariol and alternariol monomethyl, ether in fruit juices and beverages. J. Chromatogr. A 2003, 998, 119–131.
179.Ito, R.; Yamazaki, H.; Inoue, K.; Yoshimura, Y.; Kawaguchi, M.; Nakazawa, H. Development of liquid chromatography-electrospray mass spectrometry for the determination of patulin in apple juice: Investigation of its contamination levels in Japan. J. Agric. Food Chem. 2004, 52, 7464–7468.
180.Rychlik, M.; Schieberle, P. Synthesis of C-13-labeled patulin [4-hydroxy-4H-furo[3,2-c]pyran- 2(6H)-one] to be used as internal standard in a stable isotope dilution assay. J. Agric. Food Chem. 1998, 46, 5163–5169.
181.Takino, M.; Daishima, S.; Nakahara, T. Liquid chromatography/mass spectrometric determination of patulin in apple juice using atmospheric pressure photoionization. Rapid Comm. Mass Spectrom. 2003, 17, 1965–1972.
182.Krska, R.; Stubbings, G.; Macarthur, R.; Crews, C. Simultaneous determination of six major ergot alkaloids and their epimers in cereals and foodstuffs by LC-MS-MS. Anal. Bioanal. Chem. 2008, 391, 563–576.
183.Losito, I.; Monaci, L.; Aresta, A.; Zambonin, C.G. LC-ion trap electrospray MS-MS for the determination of cyclopiazonic acid in milk samples. Analyst 2002, 127, 499–502.
184.Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Comm. Mass Spectrom. 2006, 20, 2649–2659.
185.Rychlik, M.; Asam, S. Stable isotope dilution assays in mycotoxin analysis. Anal. Bioanal. Chem. 2008, 390, 617–628.
186.Stokvis, E.; Rosing, H.; Beijnen, J.H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid. Commun. Mass. Sp. 2005, 19, 401–407.
187.Kokkonen, M.; Jestoi, M.; Rizzo, A. Determination of selected mycotoxins in mould cheeses with liquid chromatography coupled to tandem with mass spectrometry. Food Addit. Contam. 2005, 22, 449–456.
188.Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64.
189.Nielsen, K.F.; Smedsgaard, J. Fungal metabolite screening: Database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography-UV-mass spectrometry methodology. J. Chromatogr. A 2003, 1002, 111–136.
190. Sibanda, L.; De Saeger, S.; Van Peteghem, C. Optimization of solid-phase clean-up prior to liquid chromatographic analysis of ochratoxin A in roasted coffee. J. Chromatogr. A 2002, 959, 327–330.
191.Aresta, A.; Vatinno, R.; Palmisano, F.; Zambonin, C.G. Determination of Ochratoxin A in wine at sub ng/mL levels by solid-phase microextraction coupled to liquid chromatography with fluorescence detection. J. Chromatogr. A 2006, 1115, 196–201.
192.Serra, R.; Mendonça, C.; Abrunhosa, L.; Pietri, A.; Venancio, A. Determination of ochratoxin A in wine grapes: Comparison of extraction procedures and method validation. Anal. Chim. Acta 2004, 513, 41–47.
193.Visconti, A.; Pascale, M.; Centonze, G. Determination of ochratoxin A in wine by means of immunoaffinity column clean-up and high-performance liquid chromatography. J. Chromatogr. A 1999, 864, 89–101.
194.Blesa, J.; Berrada, H.; Soriano, J.M.; Moltó, J.C.; Mañes, J. Rapid determination of ochratoxin A in cereals and cereal products by liquid chromatography. J. Chromatogr. A 2004, 1046, 127–131.
195.González-Peñas, E.; Leache, C.; López de Cerain, A.; Lizarraga, E. Comparison between capillary electrophoresis and HPLC-FL for ochratoxin A quantification in wine. Food Chem. 2006, 97, 349–354.
196.Jornet, D.; Busto, O.; Guasch, J. Solid-phase extraction applied to the determination of ochratoxin A in wines by reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2000, 882, 29–35.
197.Brera, C.; Grossi, S.; De Santis, B.; Miraglia, M. Automated HPLC method for the determination of Ochratoxin A in wine samples. J. Liq. Chrom. Rel. Techn. 2003, 26, 119–133.
198.Maragos, C.M.; Appell, M.; Lippolis, V.; Visconti, A.; Catucci, L.; Pascale, M. Use of cyclodextrins as modifiers of fluorescence in the detection of mycotoxins. Food Addit. Contam. 2008, 25, 164–171.
199.Varelis, P.; Leong, S.L.L.; Hocking, A.; Giannikopoulos, G. Quantitative analysis of ochratoxin A in wine and beer using solid phase extraction and high performance liquid chromatographyfluorescence detection. Food Addit. Contam. 2006, 23, 1308–1315.
200. Aboul-Enein, H.Y.; Kutluk, O.B.; Altiokka, G.; Tuncel, M. A modified HPLC method for the determination of ochratoxin A by fluorescence detection. Biomed. Chromatogr. 2002, 16, 470–474.
201.Boudra, H.; Morgavi, D.P. Development and validation of a HPLC method for the quantitation of ochratoxins in plasma and raw milk. J. Chromatogr. B 2006, 843, 295–301.
202.Curtui, V.G.; Gareis, M. A simple HPLC method for the determination of the mycotoxins ochratoxin A and B in blood serum of swine. Food Addit. Contam. 2001, 18, 635–643.
203.Hernández Hierro, J.M.; Garcia-Villanova, R.J.; Rodríguez Torrero, P.; Toruño Fonseca, I.M. Aflatoxins and ochratoxin a in red paprika for retail sale in Spain: Occurrence and evaluation of a simultaneous analytical method. J. Agric. Food Chem. 2008, 56, 751–756.
204.D'Ovidio, K.; Trucksess, M.; Weaver, C.; Horn, E.; McIntosh, M.; Bean, G. Aflatoxins in ginseng roots. Food Addit. Contam. 2006, 23, 174–180.
205.Muscarella, M.; Lo Magro, S.; Palermo, C.; Centonze, D. Validation according to European Commission Decision 2002/657/EC of a confirmatory method for aflatoxin M-1 in milk based on immunoaffinity columns and high performance liquid chromatography with fluorescence detection. Anal. Chim. Acta 2007, 594, 257–264.
206.Daradimos, E.; Marcaki, P.; Koupparis, M. Evaluation and validation of two fluorometric HPLC methods for the determination of aflatoxin B-1 in olive oil. Food Addit. Contam. 2000, 17, 65–73.
207. Pagliuca, G.; Zironi, E.; Ceccolini, A.; Matera, R.; Serrazanetti, G.P.; Piva, A. Simple method for the simultaneous isolation and determination of fumonisin B-1 and its metabolite aminopentol-1 in swine liver by liquid chromatography-fluorescence detection. J. Chromatogr. B 2005, 819, 97–103.
208.Duncan, K.; Kruger, S.; Zabe, N.; Kohn, B.; Prioli, R. Improved fluorometric and chromatographic methods for the quantification of fumonisins B-1, B-2 and B-3. J. Chromatogr. A 1998, 815, 41–47.
209.Mateo, J.J.; Mateo, R.; Hinojo, M.J.; Llorens, A.; Jiménez, M. Liquid chromatographic determination of toxigenic secondary metabolites produced by Fusarium strains. J. Chromatogr. A 2002, 955, 245–256.
210.Dilkin, P.; Mallmann, C.A.; de Almeida, C.A.A.; Corrêa, B. Robotic automated clean-up for detection of fumonisins B-1 and B-2 in corn and corn-based feed by high-performance liquid chromatography. J. Chromatogr. A 2001, 925, 151–157.
211.Stroka, J.; Capelletti, C.; Papadopoulou-Bouraoui, A.; Pallaroni, L.; Anklam, E. Investigation of alternative reagents to 2-mercaptoethanol for the pre-column derivatization of fumonisins with ophthaldialdehyde, for HPLC analysis. J. Liq. Chrom. Rel. Technol. 2002, 25, 1821–1833.
212.Lino, C.M.; Silva, L.J.G.; Pena, A.L.S.; Silveira, M.I. Determination of fumonisins B1 and B2 in Portuguese maize and maize-based samples by HPLC with fluorescence detection. Anal. Bioanal. Chem. 2006, 384, 1214–1220.
213.Jiménez, M.; Mateo, J.J.; Mateo, R. Determination of type A trichothecenes by high-performance liquid chromatography with coumarin-3-carbonyl chloride derivatisation and fluorescence detection. J. Chromatogr. A 2000, 870, 473–481.
214.Dall'Asta, C.; Galaverna, G.; Biancardi, A.; Gasparini, M.; Sforza, S.; Dossena, A.; Marchelli, R. Simultaneous liquid chromatography-fluorescence analysis of type A and type B trichothecenes as fluorescent derivatives via reaction with coumarin-3-carbonyl chloride. J. Chromatogr. A 2004, 1047, 241–247.
215.Mateo, J.J.; Llorens, A.; Mateo, R.; Jiménez, M. Critical study of and improvements in chromatographic methods for the analysis of type B trichothecenes. J. Chromatogr. A 2001, 918, 99–112.
216.Pussemier, L.; Pierard, J.Y.; Anselme, M.; Tangni, E.K.; Motte, J.C.; Larondelle, Y. Development and application of analytical methods for the determination of mycotoxins in organic and conventional wheat. Food Addit. Contam. 2006, 23, 1208–1218.
217. de Aldana, B.R.V.; Zabalgogeazcoa, I.; Ciudad, A.G.; Criado, B.G. Ergovaline occurrence in grasses infected by fungal endophytes of semi-arid pastures in Spain. J. Sci. Food Agric. 2003, 83, 347–353.
218.Jaussaud, P.; Durix, A.; Videmann, B.; Vigié, A.; Bony, S. Rapid analysis of ergovaline in ovine plasma using high-performance liquid chromatography with fluorimetric detection. J. Chromatogr. A 1998, 815, 147–153.
219.Durix, A.; Jaussaud, P.; Garcia, P.; Bonnaire, Y.; Bony, S. Analysis of ergovaline in milk using high-performance liquid chromatography with fluorimetric detection. J. Chromatogr. B 1999, 729, 255–263.
220.Cahill, L.M.; Kruger, S.C.; McAlice, B.T.; Ramsey, C.S.; Prioli, R.; Kohn, B. Quantification of deoxynivalenol in wheat using an immunoaffinity column and liquid chromatography. J. Chromatogr. A 1999, 859, 23–28.
221.Abdel-Aal, E.S.M.; Miah, K.; Young, J.C.; Rabalski, I. Comparison of four c18 columns for the liquid chromatographic determination of deoxynivalenol in naturally contaminated wheat. J. AOAC Intern. 2007, 90, 995–999.
222.Klotzel, M.; Schmidt, S.; Lauber, U.; Thielert, G.; Humpf, H.U. Comparison of different clean-up procedures for the analysis of deoxynivalenol in cereal-based food and validation of a reliable HPLC method. Chromatographia 2005, 62, 41–48.
223.Walker, F.; Meier, B. Determination of the Fusarium mycotoxins nivalenol, deoxynivalenol, 3- acetyldeoxynivalenol, and 15-O-acetyl-4-deoxynivalenol in contaminated whole wheat flour by liquid chromatography with diode array detection and gas chromatography with electron capture detection. J. AOAC Intern. 1998, 81, 741–748.
224.Llorens, A.; Mateo, R.; Mateo, J.J.; Jiménez, M. Comparison of extraction and clean-up procedures for analysis of zearalenone in corn, rice and wheat grains by high-performance liquid chromatography with photodiode array and fluorescence detection. Food Addit. Contam. 2002, 19, 272–281.
225.Wang, J.; Zhou, Y.; Wang, Q. Analysis of mycotoxin fumonisins in corn products by highperformance liquid chromatography coupled with evaporative light scattering detection. Food Chem. 2008, 107, 970–976.
226.Soleas,G.J.; Yan, J.; Goldberg, D.M. Assay of ochratoxin A in wine and beer by high-pressure liquid chromatography photodiode array and gas chromatography mass selective detection. J. Agric. Food Chem. 2001, 49, 2733–2740.
227.Katerere, D.R.; Stockenström, S.; Shephard, G.S. HPLC-DAD method for the determination of patulin in dried apple rings. Food Control 2008, 19, 389–392.
228.Gökmen, V.; Acar, J. Simultaneous determination of 5-hydroxymethylfurfural and patulin in apple juice by reversed-phase liquid chromatography. J. Chromatogr. A 1999, 847, 69–74.
229.Monaci, L.; Aresta, A.; Palmisano, F.; Visconti, A.; Zambonin, C.G. Amino-bonded silica as stationary phase for liquid chromatographic determination of cyclopiazonic acid in fungal extracts. J. Chromatogr. A 2002, 955, 79–86.
230.Solfrizzo, M.; De Girolamo, A.; Vitti, C.; Visconti, A.; van den Bulk, R. Liquid chromatographic determination of Alternaria toxins in carrots. J. AOAC Intern. 2004, 87, 101–106.
231.Tanaka, T.; Yoneda, A.; Inoue, S.; Sugiura, Y.; Ueno, Y. Simultaneous determination of trichothecene mycotoxins and zearalenone in cereals by gas chromatography-mass spectrometry. J. Chromatogr. A 2000, 882, 23–28.
232.Schothorst, R.C.; Jekel, A.A. Determination of trichothecenes in wheat by capillary gas chromatography with flame ionisation detection. Food Chem. 2001, 73, 111–117.
233.Schollenberger, M.; Lauber, U.; Terry Jara, H.; Suchy, S.; Drochner, W.; Müller, H.-M. Determination of eight trichothecenes by gas chromatography mass spectrometry after sample clean-up by a two-stage solid-phase extraction. J. Chromatogr. A 1998, 815, 123–132.
234.Jestoi, M.; Ritieni, A.; Rizzo, A. Analysis of the Fusarium mycotoxins fusaproliferin and trichothecenes in grains using gas chromatography-mass spectrometry. J. Agric. Food Chem. 2004, 52, 1464–1469.
235.Rodrigues-Fo, E.; Mirocha, C.J.; Xie, W.P.; Krick, T.P.; Martinelli, J.A. Electron ionization mass spectral fragmentation of deoxynivalenol and related tricothecenes. Rapid Commun. Mass Spectrom. 2002, 16, 1827–1835.
236.Rupp, H.S.; Turnipseed, S.B. Confirmation of patulin and 5-hydroxymethylfurfural in apple juice by gas chromatography/mass spectrometry. J. AOAC Intern. 2000, 83, 612–620.
237.Shu, P.Y.; Lin, C.H. Simple and sensitive determination of citrinin in Monascus by GC-selected ion monitoring mass spectrometry. Anal. Sci. 2002, 18, 283–287.
238.Stroka, J.; van Otterdijk, R.; Anklam, E. Immunoaffinity column clean-up prior to thin-layer chromatography for the determination of aflatoxins in various food matrices. J. Chromatogr. A 2000, 904, 251–256.
239.Otta, K.H.; Papp, E.; Mincsovics, E.; Záray, G. Determination of aflatoxins in corn by use of the personal OPLC basic system. JPC – J. Plan. Chromatogr. – Modern TLC 1998, 11, 370–373.
240.Papp, E.; Otta, K.H.; Záray, G.; Mincsovics, E. Liquid chromatographic determination of aflatoxins. Microchem. J. 2002, 73, 39–46.
241.Stroka, J.; Dasko, L.; Spangenberg, B.; Anklam, E. Determination of the mycotoxin, sterigmatocystin, by thin-layer chromatography and reagent-free derivatisation. J. Liq. Chrom. Rel. Technol. 2004, 27, 2101–2111.
242.Ostry, V.; Skarkova, J. Development of an HPTLC method for the determination of deoxynivalenol in cereal products. JPC – J. Plan. Chromatogr. – Modern TLC 2000, 13, 443– 446.
243.Dawlatana, M.; Coker, R.D.; Nagler, M.J.; Gibbs, J. Blunden, G. An HPTLC method for the quantitative determination of T-2 toxin and deoxynivalenol in rice. Chromatographia 1999, 49, 547–551.
244.Preis, R.A.; Vargas, E.A. A method for determining fumonisin B-1 in corn using immunoaffinity column clean-up and thin layer chromatography/densitometry. Food Addit. Contam. 2000, 17, 463–468.
245.Karuna, R.; Sashidhar, R.B. Use of ion-exchange chromatography coupled with TLC-laser scanning densitometry for the quantitation of fumonisin B-1. Talanta 1999, 50, 381–389.
246.Hadiani, M.R.; Yazdanpanah, H.; Ghazi-Khansari, M.; Cheraghali, A.M.; Goodarzi, M. Survey of the natural occurrence of zearalenone in maize from northern Iran by thin-layer chromatography densitometry. Food Addit. Contam. 2003, 20, 380–385.
247.Dawlatana, M.; Coker, R.D.; Nagler, M.J.; Blunden, G.; Oliver, G.W.O. An HPTLC method for the quantitative determination of zearalenone in maize. Chromatographia 1998, 47, 215–218.
248.Sørensen, J.L.; Nielsen, K.F.; Thrane, U. Analysis of moniliformin in maize plants using hydrophilic interaction chromatography. J. Agric. Food Chem. 2007, 55, 9764–9768.
249.Apfelthaler, E.; Bicker, W.; Lammerhofer, M.; Sulyok, M.; Krska, R.; Lindner, W.; Schuhmacher, R. Retention pattern profiling of fungal metabolites on mixed-mode reversed-phase/weak anion exchange stationary phases in comparison to reversed-phase and weak anion exchange separation materials by liquid chromatography-electrospray ionisation-tandem mass spectrometry. J. Chromatogr. A 2008, 1191, 171–181.
250.Hu, D.J.; Liu, M.; Xia, X.; Chen, D.J.; Zhao, F.S.; Ge, M. Preparative isolation and purification of Altertoxin I from an Alternaria sp By HSCCC. Chromatographia 2008, 67, 863–867.
251.Dalsgaard, P.W.; Nielsen, K.F.; Larsen, T.O. UV-guided isolation of fungal metabolites by HSCCC. J. Liq. Chrom. Rel. Technol. 2005, 28, 2029–2039.
252.Corneli, S.; Maragos, C.M. Capillary electrophoresis with laser-induced fluorescence: Method for the mycotoxin ochratoxin A. J. Agric. Food Chem. 1998, 46, 3162–3165.
253.Köller, G.; Rolle-Kampczyk, U.; Popp, P.; Herbarth, O. A new method for the sensitive analysis of Ochratoxin A in house dust. Chromatographia 2003, 58, 763–767.
254.Köller, G.; Rolle-Kampczyk, U.; Lehmann, I.; Popp, P.; Herbarth, O. Determination of Ochratoxin A in small volumes of human blood serum. J. Chromatogr. B 2004, 804, 313–317.
255.Maragos, C.M.; Appell, M. Capillary electrophoresis of the mycotoxin zearalenone using cyclodextrin-enhanced fluorescence. J. Chromatogr. A 2007, 1143, 252–257.
256.Murillo, M.; González-Peñas, E.; Amézqueta, S. Determination of patulin in commercial apple juice by micellar electrokinetic chromatography. Food Chem. Toxicol. 2008, 46, 57–64.
257.Murillo-Arbizu, M.; González-Peñas, E.; Hansen, S.H.; Amézqueta, S.; Østergaard, J. Development and validation of a microemulsion electrokinetic chromatography method for patulin quantification in commercial apple juice. Food Chem. Toxicol. 2008, 46, 2251–2257.
258.Molina, P.G.; Zón, M.A.; Fernández, H. The electrochemical behaviour of the altenuene mycotoxin and its acidic properties. J. Electroanal. Chem. 2002, 520, 94–100.
259.Sheibani, A.; Tabrizchi, M.; Ghaziaskar, H.S. Determination of aflatoxins B1 and B2 using ion mobility spectrometry. Talanta 2008, 75, 233–238.
260.McCooeye, M.; Kolakowski, B.; Boison, J.; Mester, Z. Evaluation of high-field asymmetric waveform ion mobility spectrometry mass spectrometry for the analysis of the mycotoxin zearalenone. Anal. Chim. Acta 2008, 627, 112–116.
261. Anselme, M.; Tangni, E.K.; Pussemier, L.; Motte, J.-C.; Van Hove, F.; Schneider, Y.-J.; Van Peteghem, C.; Larondelle, Y. Comparison of ochratoxin A and deoxynivalenol in organically and conventionally produced beers sold on the Belgian market. Food Addit. Contam. 2006, 23, 910–918.
262.Belli, N.; Marin, S.; Sanchis, V.; Ramos, A.J. Review: Ochratoxin A (OTA) in wines, musts and grape juices: Occurrence, regulations and methods of analysis. Food Sci. Technol. Inern. 2002, 8, 325–335.
263.Baert, K.; De Meulenaer, B.; Kasase, C.; Huyghebaert, A.; Ooghe, W.; Devlieghere, F. Free and bound patulin in cloudy apple juice. Food Chem. 2007, 100, 1278–1282.
264. Molinié, A.; Faucet, V.; Castegnaro, M.; Pfohl-Leszkowicz, A. Analysis of some breakfast cereals on the French market for their contents of ochratoxin A, citrinin and fumonisin B-1: Development of a method for simultaneous extraction of ochratoxin A and citrinin. Food Chem. 2005, 92, 391–400.
265.Solfrizzo, M.; Avantaggiato, G.; Visconti, A. Use of various clean-up procedures for the analysis of ochratoxin A in cereals. J. Chromatogr. A 1998, 815, 67–73.
266.Krska, R.; Baumgartner, S.; Josephs, R. The state-of-the-art in the analysis of type-A and -B trichothecene mycotoxins in cereals. Fresenius' J. Anal. Chem. 2001, 371, 285–299.
267.De Girolamo, A.; Solfrizzo, M.; von Holst, C.; Visconti, A. Comparison of different extraction and clean-up procedures for the determination of fumonisins in maize and maize-based food products. Food Addit. Contam. 2001, 18, 59–67.
268.Tanaka, K.; Sago, Y.; Zheng, Y.; Nakagawa, H.; Kushiro, M. Mycotoxins in rice. Intern. J. Food Microbiol. 2007, 119, 59–66.
269.Nguyen, M.T.; Tozovanu, M.; Tran, T.L.; Pfohl-Leszkowicz, A. Occurrence of aflatoxin B1, citrinin and ochratoxin A in rice in five provinces of the central region of Vietnam. Food Chem. 2007, 105, 42–47.
270.Schollenberger, M.; Müller, H.-M.; Rüfle, M.; Terry-Jara, H.; Suchy, S.; Plank, S.; Drochner, W. Natural occurrence of Fusarium toxins in soy food marketed in Germany. Int. J. Food. Microbiol. 2007, 113, 142–146.
271.Bonvehi, J.S. Occurrence of ochratoxin A in cocoa products and chocolate. J. Agric. Food Chem. 2004, 52, 6374–6352.
272.Sanchez-Hervas, M.; Gil, J.V.; Bisbal, F.; Ramon, D.; Martinez-Culebras, P.V. Mycobiota and mycotoxin producing fungi from cocoa beans. Intern. J. Food Microbiol. 2008, 125, 336–340.
273.Brera, C.; Grossi, S.; Miraglia, M. Interlaboratory study for ochratoxin a determination in cocoa powder samples. J. Liq. Chrom. Rel. Technol. 2005, 28, 35–61.
274.Sypecka, Z.; Kelly, M.; Brereton, P. Deoxynivalenol and zearalenone residues in eggs of laying hens fed with a naturally contaminated diet: Effects on egg production and estimation of transmission rates from feed to eggs. J. Agric. Food Chem. 2004, 52, 5463–5471.
275.Oliveira, C.A.F.; Kobashigawa, E.; Reis, T.A.; Mestieri, L.; Albaquerque, R.; Correa, B. Aflatoxin B-1 residues in eggs of laying hens fed a diet containing different levels of the mycotoxin. Food Addit. Contam. 2000, 17, 459–462.
276.Prandini, A.; Tansini, G.; Sigolo, S.; Filippi, L.; Laporta, M.; Piva, G. On the occurrence of aflatoxin M1 in milk and dairy products. Food Chem. Toxicol. 2008, DOI: 10.1016/j.fct.2007.10.005.
277.Josephs, R.D.; Ulberth, F.; Van Egmond, H.P.; Emons, H. Aflatoxin M-1 in milk powders: Processing, homogeneity and stability testing of certified reference materials. Food Addit. Contam. 2005, 22, 864–874.
278.Govaris, A.; Roussi, V.; Koidis, P.A.; Botsoglou, N.A. Distribution and stability of aflatoxin M-1 during production and storage of yoghurt. Food Addit. Contam. 2002, 19, 1043–1050.
279.Meky, F.A.; Turner, P.C.; Ashcroft, A.E.; Miller, J.D.; Qiao, Y.-L.; Roth, M.J.; Wild, C.P. Development of a urinary biomarker of human exposure to deoxynivalenol. Food Chem. Toxicol. 2003, 41, 265–273.
280.Tor, E.R.; Puschner. B.; Filigenzi, M.S.; Tiwary, A.K.; Poppenga, R.H. LC-MS/MS screen for penitrem A and roquefortine C in serum and urine samples. Anal. Chem. 2006, 78, 4624–4629.
281.Yang, Z.; Wang, S.H. Recent development in application of high performance liquid chromatography-tandem mass spectrometry in therapeutic drug monitoring of immunosuppressants. J. Immunol. Methods 2008, 336, 98–103.
282.Meyer, K.; Mohr, K.; Bauer, J.; Horn, P.; Kovacs, M. Residue formation of fumonisin B1 in porcine tissues. Food Addit. Contam. 2003, 20, 639–647.
283.Brera, C.; Miraglia, M.; Colatosti, M. Evaluation of the impact of mycotoxins on human health: Sources of errors. Microchem. J. 1998, 59, 45–49.
284.Edberg Hansen, M.; Smedsgaard, J.; Ostenfeld Larsen, T. X-hitting: An algorithm for novelty detection and dereplication by UV spectra of complex mixtures of natural products. Anal. Chem. 2005, 77, 6805–6817.
285.Hirota, M.; Menta, A.B.; Yoneyama, K.; Kitabatake, N. A major decomposition product, citrinin H2, from citrinin on heating with moisture. Biosci. Biotechn. Biochem. 2002, 66, 206–210.
286.Kinani, S.; Bouchonnet, S.; Bourcier, S.; Porcher, J.-M.; Aït-Aïssa, S. Study of the chemical derivatization of zearalenone and its metabolites for gas chromatography-mass spectrometry analysis of environmental samples. J. Chromatogr. A 2008, 1190, 307–315.
287.Pena, A.; Seifrtová, M.; Lino, C.; Silveira, I.; Solich, P. Estimation of ochratoxin A in portuguese population: New data on the occurrence in human urine by high performance liquid chromatography with fluorescence detection. Food Chem. Toxicol. 2006, 44, 1449–1454.
288.Whitaker, T.B. Standardisation of mycotoxin sampling procedures: An urgent necessity. Food Control 2003, 14, 233–237.
289.Whitaker, T.B.; Hagler, W.M.; Giesbrecht, F.G.; Johansson, A.S. Sampling, sample preparation, and analytical variability associated with testing wheat for deoxynivalenol. J. AOAC Intern. 2000, 83, 1285–1292.
290.Stroka, J.; Spanjer, M.; Buechler, S.; Barel, S.; Kos, G.; Anklam, E. Novel sampling methods for the analysis of mycotoxins and the combination with spectroscopic methods for the rapid evaluation of deoxynivalenol contamination. Toxicol. Lett. 2004, 153, 99–107.
291.Champeil, A.; Fourbet, J.-F.; Doré, T. Effects of grain sampling procedures on Fusarium mycotoxin assays in wheat grains. J. Agric. Food Chem. 2004, 52, 6049–6054.
292.Spanjer, M.C.; Scholten, J.M.; Kastrup, S.; Jorissen, U.; Schatzki, T.F.; Toyofuku, N. Sample comminution for mycotoxin analysis: Dry milling or slurry mixing? Food Addit. Contam. 2006, 23, 73–83.
293.Visconti, A.; De Girolamo, A. Fitness for purpose - Ochratoxin A analytical developments. Food Addit. Contam. 2005, 22 (Suppl. 1), 37–44.
294.Josephs, R.D.; Derbyshire, M.; Stroka, J.; Emons, H.; Anklam, E. Trichothecenes: Reference materials and method validation. Toxicol. Lett. 2004, 153, 123–132.
295.Krska, R.; Schothorst, R.C.; van Egmond, H.P.; Josephs, R.D.; Lepschy, J.; Pettersson, H.; Chan, D.; Berthiller, F.; Schuhmacher, R.; Kandler, W.; Parich, A.; Welzig, E. Processing and purity assessment of standards for the analysis of type-B trichothecene mycotoxins. Anal. Bioanal. Chem. 2005, 382, 1848–1858.
296.Krska, R.; Welzig, E.; Berthiller, F.; Molinelli, A.; Mizaikoff, B. Advances in the analysis of mycotoxins and its quality assurance. Food Addit. Contam. 2005, 22, 345–353.
297.Krska, R.; Welzig, E.; Drs, E.; Josephs, R.D.; Schothorst, R.C.; van Egmond, H.P.; Pettersson, H.; Chan, D.; MacDonald, S. Feasibility study for the production of certified calibrants for the determination of deoxynivalenol and other B-trichothecenes: Intercomparison study. J. AOAC Intern. 2006, 89, 1573–1580.
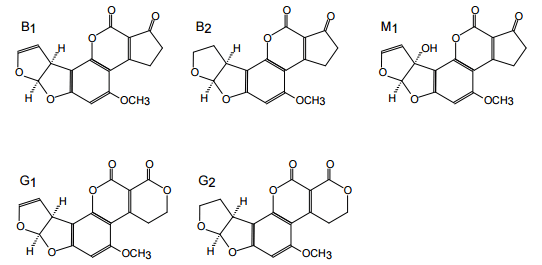
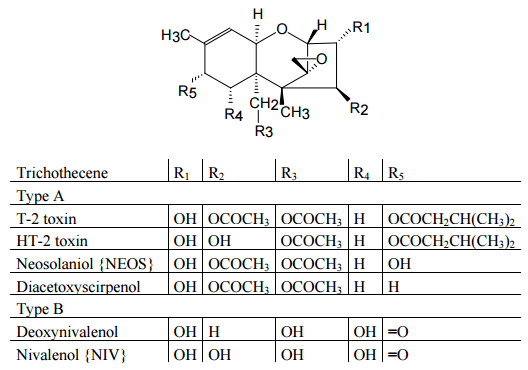
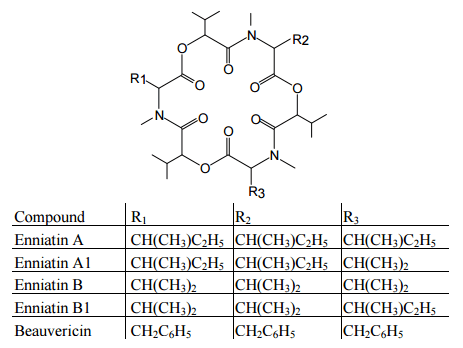
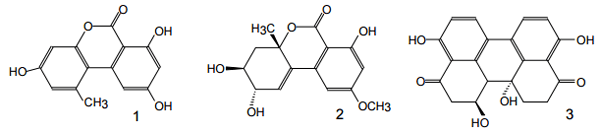
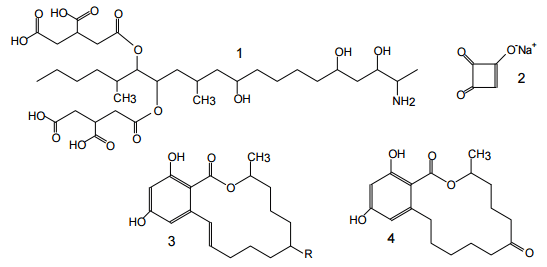
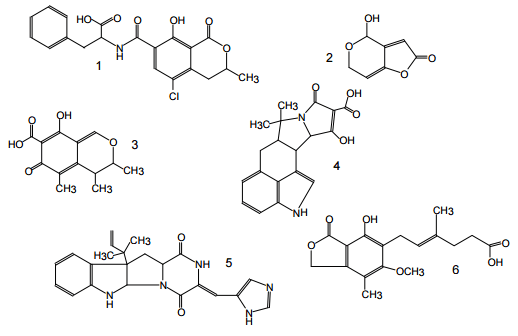
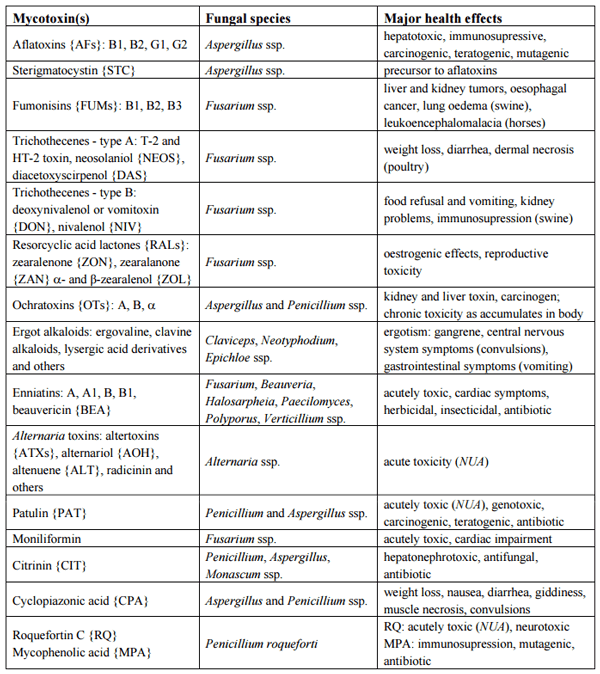
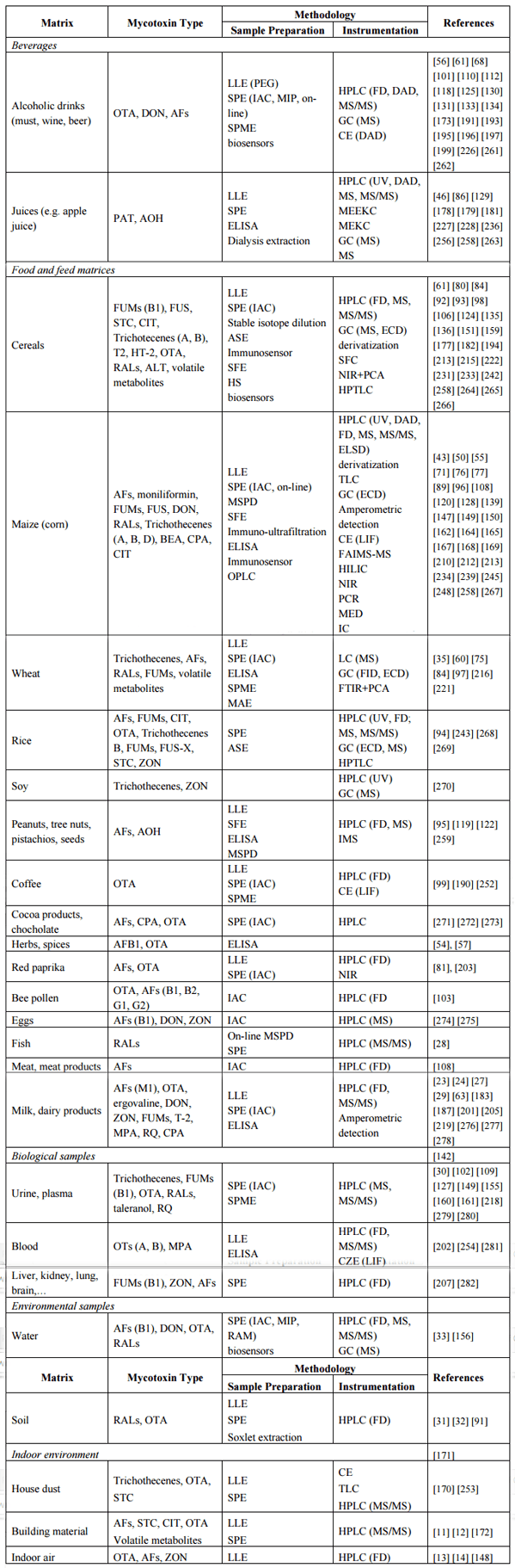








.jpg&w=3840&q=75)