Effects of Eimeria maxima and Clostridium perfringens infections on cecal microbial composition and the possible correlation with body weight gain in broiler chickens
Author details:
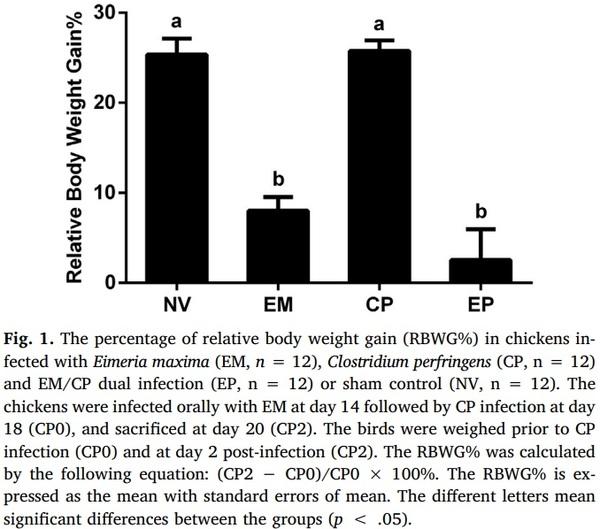
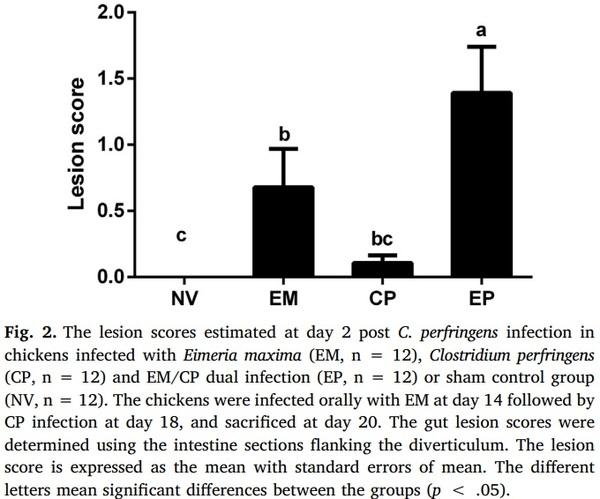
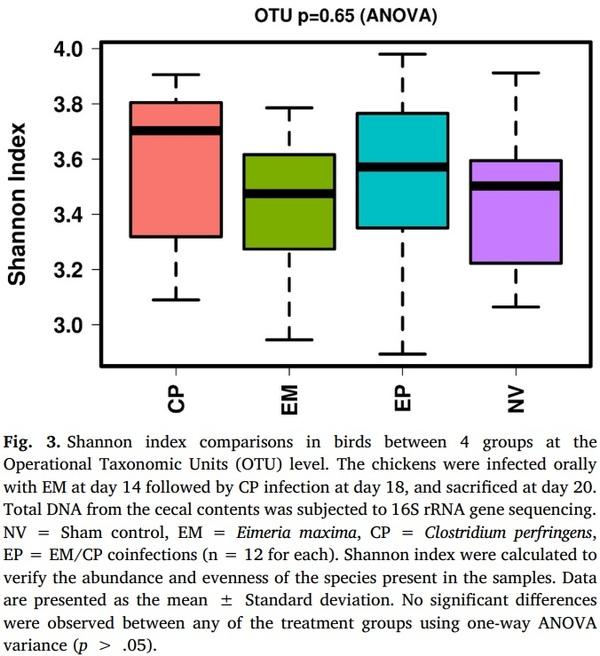
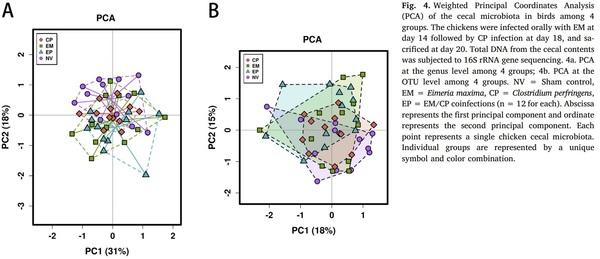

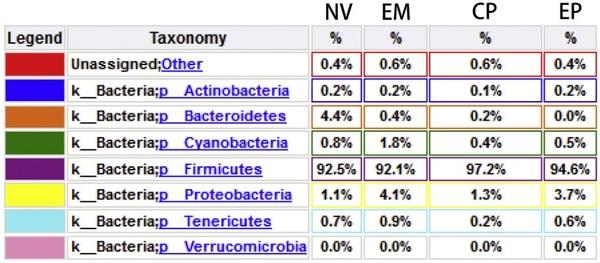
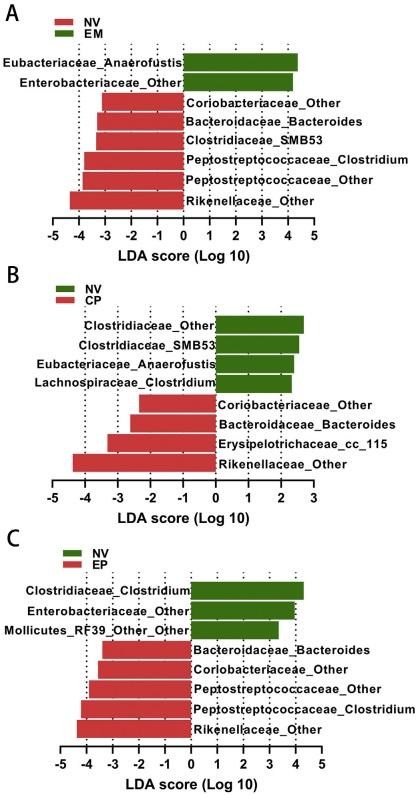
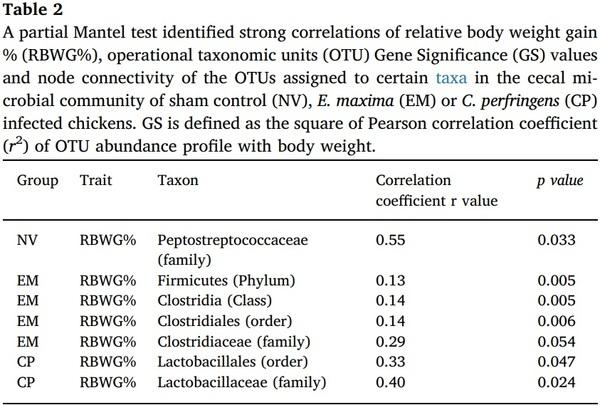
Aleksandrzak-Piekarczyk, T., Puzia, W., Zylinska, J., Ciesla, J., Gulewicz, K.A.,
Bardowski, J.K., Gorecki, R.K., 2019. Potential of Lactobacillus plantarum IBB3036 and Lactobacillus salivarius IBB3154 to persistence in chicken after in ovo delivery.
Microbiologyopen 8, e00620.
Angelakis, E., 2017. Weight gain by gut microbiota manipulation in productive animals.
Microb. Pathog. 106, 162–170.
Ayyanna, R., Ankaiah, D., Arul, V., 2018. Anti-inflammatory and antioxidant properties of probiotic bacterium Lactobacillus mucosae AN1 and Lactobacillus fermentum SNR1 in wistar albino rats. Front. Microbiol. 9, 3063.
Bauer, M.A., Kainz, K., Carmona-Gutierrez, D., Madeo, F., 2018. Microbial wars: competition in ecological niches and within the microbiome. Microbial Cell 5, 215.
Belizario, J.E., Faintuch, J., Garay-Malpartida, M., 2018. Gut microbiome dysbiosis and immunometabolism: new frontiers for treatment of metabolic diseases. Mediat.
Inflamm. 2018, 2037838.
Bermingham, E.N., Maclean, P., Thomas, D.G., Cave, N.J., Young, W., 2017. Key bacterial families (Clostridiaceae, Erysipelotrichaceae and Bacteroidaceae) are related to the digestion of protein and energy in dogs. PeerJ 5, e3019.
Bortoluzzi, C., Vieira, B.S., Hofacre, C., Applegate, T.J., 2019. Effect of different challenge models to induce necrotic enteritis on the growth performance and intestinal microbiota of broiler chickens. Poult. Sci. 98, 2800–2812.
Caly, D.L., D’Inca, R., Auclair, E., Drider, D., 2015. Alternatives to antibiotics to prevent necrotic enteritis in broiler chickens: a microbiologist’s perspective. Front. Microbiol.
6, 1336.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K.,
Fierer, N., Pena, A.G., Goodrich, J.K., Gordon, J.I., 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335.
Caterson, B., Melrose, J., 2018. Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology 28, 182–206.
Chapman, H.D., 2014. Milestones in avian coccidiosis research: a review. Poult. Sci. 93,
501–511.
Chen, C., Chuang, L., Chiang, Y., Lin, C., Lien, Y., Tsean, H., 2016. Use of a probiotic to ameliorate the growth rate and the inflammation of broiler chickens caused by
Eimeria tenella infection. J. Anim. Res. Nutr. 1.
Cho, Y.J., Choi, J.K., Kim, J.H., Lim, Y.S., Ham, J.S., Kang, D.K., Chun, J., Paik, H.D., Kim,
G.B., 2011. Genome sequence of Lactobacillus salivarius GJ-24, a probiotic strain isolated from healthy adult intestine. J. Bacteriol. 193, 5021–5022.
Clarke, S.F., Murphy, E.F., O'Sullivan, O., Ross, R.P., O'Toole, P.W., Shanahan, F., Cotter,
P.D., 2013. Targeting the microbiota to address diet-induced obesity: a time dependent challenge. PLoS One 8, e65790.
Clavijo, V., Florez, M.J.V., 2018. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: a review. Poult. Sci. 97,
1006–1021.
Collier, C.T., Hofacre, C.L., Payne, A.M., Anderson, D.B., Kaiser, P., Mackie, R.I., Gaskins,
H.R., 2008. Coccidia-induced mucogenesis promotes the onset of necrotic enteritis by supporting Clostridium perfringens growth. Vet. Immunol. Immunopathol. 122,
104–115.
Cooper, K.K., Songer, J.G., 2010. Virulence of Clostridium perfringens in an experimental model of poultry necrotic enteritis. Vet. Microbiol. 142, 323–328.
Corr, S.C., Li, Y., Riedel, C.U., O’Toole, P.W., Hill, C., Gahan, C.G., 2007. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius
UCC118. Proc. Natl. Acad. Sci. U. S. A. 104, 7617–7621.
Fakhry, S., Manzo, N., D’Apuzzo, E., Pietrini, L., Sorrentini, I., Ricca, E., De Felice, M.,
Baccigalupi, L., 2009. Characterization of intestinal bacteria tightly bound to the human ileal epithelium. Res. Microbiol. 160, 817–823.
Fath, M.J., Kolter, R., 1993. ABC transporters: bacterial exporters. Microbiol. Mol. Biol.
Rev. 57, 995–1017.
Gharib-Naseri, K., Kheravii, S.K., Keerqin, C., Morgan, N., Swick, R.A., Choct, M., Wu,
S.B., 2019. Two different Clostridium perfringens strains produce different levels of necrotic enteritis in broiler chickens. Poult. Sci. 98, 6422–6432.
Helmann, J.D., 2002. The extracytoplasmic function (ECF) sigma factors. Adv. Microb.
Physiol. 46, 48–111.
Hessenberger, S., Schatzmayr, G., Teichmann, K., 2016. In vitro inhibition of Eimeria tenella sporozoite invasion into host cells by probiotics. Vet. Parasitol. 229, 93–98.
Hofacre, C.L., Smith, J.A., Mathis, G.F., 2018. An optimist’s view on limiting necrotic enteritis and maintaining broiler gut health and performance in today’s marketing, food safety, and regulatory climate. Poult. Sci. 97, 1929–1933.
Kaboudi, K., Umar, S., Munir, M.T., 2016. Prevalence of coccidiosis in free-range chicken in Sidi Thabet, Tunisia. Scientifica 2016.
Lacey, J.A., Stanley, D., Keyburn, A.L., Ford, M., Chen, H., Johanesen, P., Lyras, D.,
Moore, R.J., 2018. Clostridium perfringens-mediated necrotic enteritis is not influenced by the pre-existing microbiota but is promoted by large changes in the postchallenge microbiota. Vet. Microbiol. 227, 119–126.
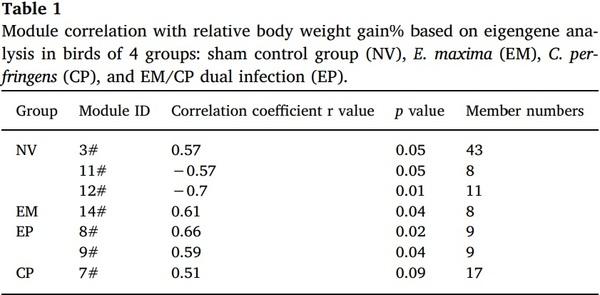
Langfelder, P., Horvath, S., 2007. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1, 54.
Latorre, J.D., Adhikari, B., Park, S.H., Teague, K.D., Graham, L.E., Mahaffey, B.D., Baxter,
M.F.A., Hernandez-Velasco, X., Kwon, Y.M., Ricke, S.C., Bielke, L.R., Hargis, B.M.,
Tellez, G., 2018. Evaluation of the epithelial barrier function and ileal microbiome in an established necrotic enteritis challenge model in broiler chickens. Front Vet Sci 5,
199.
Lee, S.H., Lillehoj, H.S., Jang, S.I., Lillehoj, E.P., Min, W., Bravo, D.M., 2013. Dietary supplementation of young broiler chickens with Capsicum and turmeric oleoresins increases resistance to necrotic enteritis. Br. J. Nutr. 110, 840–847.
Lee, K.-C., Kil, D.Y., Sul, W.J., 2017. Cecal microbiome divergence of broiler chickens by sex and body weight. J. Microbiol. 55, 939–945.
Lee, Y.S., Lee, S.H., Gadde, U.D., Oh, S.T., Lee, S.J., Lillehoj, H.S., 2018. Allium hookeri supplementation improves intestinal immune response against necrotic enteritis in young broiler chickens. Poult. Sci. 97, 1899–1908.
Li, R.W., Li, W., Sun, J., Yu, P., Baldwin, R.L., Urban, J.F., 2016. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci. Rep. 6, 20606.
Li, C., Lillehoj, H.S., Gadde, U.D., Ritter, D., Oh, S., 2017. Characterization of Clostridium perfringens strains isolated from healthy and necrotic enteritis-afflicted broiler chickens. Avian Dis. 61, 178–185.
Li, C., Yan, X., Lillehoj, H.S., Oh, S., Liu, L., Sun, Z., Gu, C., Lee, Y., Xianyu, Z., Zhao, H.,
2019. Eimeria maxima-induced transcriptional changes in the cecal mucosa of broiler chickens. Parasit. Vectors 12, 285.
Lillehoj, H.S., Jang, S.I., Panebra, A., Lillehoj, E.P., Dupuis, L., Ben Arous, J., Lee, S.K.,
Oh, S.T., 2017. In ovo vaccination using Eimeria profilin and Clostridium perfringens
NetB proteins in Montanide IMS adjuvant increases protective immunity against experimentally-induced necrotic enteritis. Asian-Australas J Anim Sci 30,
1478–1485.
Liu, F., Li, Z., Wang, X., Xue, C., Tang, Q., Li, R.W., 2019. Microbial co-occurrence patterns and keystone species in the gut microbial community of mice in response to stress and chondroitin sulfate sisaccharide. Int. J. Mol. Sci. 20.
Lo Verso, L., Lessard, M., Talbot, G., Fernandez, B., Fliss, I., 2018. Isolation and selection of potential probiotic bacteria from the pig gastrointestinal tract. Probiotics
Antimicrob Proteins 10, 299–312.
Macdonald, S.E., Nolan, M.J., Harman, K., Boulton, K., Hume, D.A., Tomley, F.M.,
Stabler, R.A., Blake, D.P., 2017. Effects of Eimeria tenella infection on chicken caecal microbiome diversity, exploring variation associated with severity of pathology. PLoS
One 12, e0184890.
Messaoudi, S., Manai, M., Kergourlay, G., Prevost, H., Connil, N., Chobert, J.M., Dousset,
X., 2013. Lactobacillus salivarius: bacteriocin and probiotic activity. Food Microbiol.
36, 296–304.
Million, M., Angelakis, E., Paul, M., Armougom, F., Leibovici, L., Raoult, D., 2012.
Comparative meta-analysis of the effect of Lactobacillus species on weight gain in humans and animals. Microb. Pathog. 53, 100–108.
Miska, K., Fetterer, R., 2016. The mRNA expression of amino acid and sugar transporters, aminopeptidase, as well as the di-and tri-peptide transporter PepT1 in the intestines of Eimeria infected broiler chickens. Poult. Sci. 96, 465–473.
Mokhtari, A., Blancato, V.S., Repizo, G.D., Henry, C., Pikis, A., Bourand, A., de Fátima
Álvarez, M., Immel, S., Mechakra-Maza, A., Hartke, A., 2013. Enterococcus faecalis utilizes maltose by connecting two incompatible metabolic routes via a novel maltose
6′-phosphate phosphatase (MapP). Mol. Microbiol. 88, 234–253.
Moore, R.J., 2016. Necrotic enteritis predisposing factors in broiler chickens. Avian
Pathol 45, 275–281.
Niccolai, E., Boem, F., Russo, E., Amedei, A., 2019. The gut(−)brain axis in the neuropsychological disease model of obesity: a classical movie revised by the emerging director “microbiome”. Nutrients 11.
Oh, S., Gadde, U.D., Bravo, D., Lillehoj, E.P., Lillehoj, H.S., 2018. Growth-promoting and antioxidant effects of magnolia bark extract in chickens uninfected or co-infected with Clostridium perfringens and Eimeria maxima as an experimental model of necrotic enteritis. Curr Dev Nutr 2 nzy009.
Paget, M.S., 2015. Bacterial sigma factors and anti-sigma factors: structure, function and distribution. Biomolecules 5, 1245–1265.
Parreira, V.R., Ojha, S., Lepp, D., Mehdizadeh Gohari, I., Zhou, H., Susta, L., Gong, J.,
Prescott, J.F., 2017. Necrotic enteritis locus 1 diguanylate cyclase and phosphodiesterase (cyclic-di-GMP) gene mutation attenuates virulence in an avian necrotic enteritis isolate of Clostridium perfringens. Vet. Microbiol. 208, 69–73.
Prescott, J.F., Smyth, J.A., Shojadoost, B., Vince, A., 2016. Experimental reproduction of necrotic enteritis in chickens: a review. Avian Pathol 45, 317–322.
Qian, L.C., Sun, J., 2009. Effect of β-glucosidase as a feed supplementary on the growth performance, digestive enzymes and physiology of broilers. Asian Australas. J. Anim.
Sci. 22, 260–266.
Rajilic-Stojanovic, M., de Vos, W.M., 2014. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 38, 996–1047.
Relman, D.A., Lipsitch, M., 2018. Microbiome as a tool and a target in the effort to address antimicrobial resistance. PNAS 115, 12902–12910.
Ritzi, M.M., Abdelrahman, W., Van-Heerden, K., Mohnl, M., Barrett, N.W., Dalloul, R.A.,
2016. Combination of probiotics and coccidiosis vaccine enhances protection against an Eimeria challenge. Vet. Res. 47, 111.
Sarson, A.J., Wang, Y., Kang, Z., Dowd, S.E., Lu, Y., Yu, H., Han, Y., Zhou, H., Gong, J.,
2009. Gene expression profiling within the spleen of Clostridium perfringens-challenged broilers fed antibiotic-medicated and non-medicated diets. BMC Genomics 10,
260.
Scriver, C.R., 2001. The metabolic & molecular bases of inherited disease. In: New York.
McGraw-Hill, Montreal.
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W.S., Huttenhower,
C., 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60.
Senok, A.C., Ismaeel, A.Y., Botta, G.A., 2005. Probiotics: facts and myths. Clin. Microbiol.
Infect. 11, 958–966.
Shirley, M.W., Ivens, A., Gruber, A., Madeira, A.M., Wan, K.L., Dear, P.H., Tomley, F.M.,
2004. The Eimeria genome projects: a sequence of events. Trends Parasitol. 20,
199–201.
Stanley, D., Keyburn, A.L., Denman, S.E., Moore, R.J., 2012. Changes in the caecal microflora of chickens following Clostridium perfringens challenge to induce necrotic enteritis. Vet. Microbiol. 159, 155–162.
Stanley, D., Wu, S.B., Rodgers, N., Swick, R.A., Moore, R.J., 2014. Differential responses of cecal microbiota to fishmeal, Eimeria and Clostridium perfringens in a necrotic enteritis challenge model in chickens. PLoS One 9, e104739.
Su, S., Miska, K., Fetterer, R., Jenkins, M., Wong, E., 2015. Expression of digestive enzymes and nutrient transporters in Eimeria-challenged broilers. Exp. Parasitol. 150,
13–21.
Wade, B., Keyburn, A., 2015. The true cost of necrotic enteritis. World Poult 31, 16–17.
Wang, Y., Liu, F., Urban Jr., J.F., Paerewijck, O., Geldhof, P., Li, R.W., 2019. Ascaris suum infection was associated with a worm-independent reduction in microbial diversity and altered metabolic potential in the porcine gut microbiome. Int. J. Parasitol. 49,
247–256.
Watanabe, M., Kinoshita, H., Nitta, M., Yukishita, R., Kawai, Y., Kimura, K., Taketomo,
N., Yamazaki, Y., Tateno, Y., Miura, K., Horii, A., Kitazawa, H., Saito, T., 2010.
Identification of a new adhesin-like protein from Lactobacillus mucosae ME-340 with specific affinity to the human blood group a and B antigens. J. Appl. Microbiol. 109,
927–935.
Wexler, H.M., 2007. Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol.
Rev. 20, 593–621.
Williams, R., 2005. Intercurrent coccidiosis and necrotic enteritis of chickens: rational, integrated disease management by maintenance of gut integrity. Avian Pathol. 34,
159–180.
Wu, S.B., Stanley, D., Rodgers, N., Swick, R.A., Moore, R.J., 2014. Two necrotic enteritis predisposing factors, dietary fishmeal and Eimeria infection, induce large changes in the caecal microbiota of broiler chickens. Vet. Microbiol. 169, 188–197.
Yegani, M., Korver, D.R., 2008. Factors affecting intestinal health in poultry. Poult. Sci.
87, 2052–2063.
Zhang, C., Li, S., Yang, L., Huang, P., Li, W., Wang, S., Zhao, G., Zhang, M., Pang, X., Yan,
Z., Liu, Y., Zhao, L., 2013. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 4, 2163.
Zhang, X., Wang, Y., Yan, Y., Peng, H., Long, Y., Zhang, Y., Jiang, Z., Liu, P., Zou, C.,
Peng, H., 2019. Transcriptome sequencing analysis of maize embryonic callus during early redifferentiation. BMC Genomics 20, 159.
Zhou, B., Yang, L., Li, S., Huang, J., Chen, H., Hou, L., Wang, J., Green, C.D., Yan, Z.,
Huang, X., Kaeberlein, M., Zhu, L., Xiao, H., Liu, Y., Han, J.D., 2012. Midlife gene expressions identify modulators of aging through dietary interventions. Proc. Natl.
Acad. Sci. U. S. A. 109, E1201–E1209.









