Transcriptome Alterations of an in vitro-Selected, Moderately Resistant, Two-Row Malting Barley in Response to 3ADON, 15ADON, and NIV Chemotypes of Fusarium graminearum
Fusarium head blight caused by Fusarium graminearum is a devastating disease of malting barley. Mycotoxins associated with contaminated grain can be transferred from malt to beer and pose a health risk to consumers. In western Canada, F. graminearum has undergone an adaptive shift from 15ADON constituency to dominance by virulent 3ADON-producers; likewise, NIV-producers have established in regions of southern United States. Lack of adapted resistance sources with adequate malting quality has promoted the use of alternative breeding methodologies, such as in vitro selection. We studied the low-deoxynivalenol characteristic of in vitro selected, two-row malting barley variety “Norman” by RNAseq in contrast to its parental line “CDC Kendall,” when infected by 15ADON-, 3ADON-, and NIV-producing isolates of F. graminearum. The current study documents higher mycotoxin accumulation by 3ADON isolates, thereby representing increased threat to barley production. At 72–96-h post infection, significant alterations in transcription patterns were observed in both varieties with pronounced upregulation of the phenylpropanoid pathway and detoxification gene categories (UGT, GST, CyP450, and ABC), particularly in 3ADON treatment. Defense response was multitiered, where differential expression in “Norman” associated with antimicrobial peptides (thionin 2.1, defensing, non-specific lipid-transfer protein) and stress-related proteins, such as late embryogenesis abundant proteins, heat-shock, desiccation related, and a peroxidase (HvPrx5). Several gene targets identified in “Norman” would be useful for application of breeding varieties with reduced deoxynivalenol content.
Keywords: malt, Fusarium head blight, Fusarium graminearum, RNA-Seq, in vitro selection, deoxynivalenol.
INTRODUCTION
MATERIALS AND METHODS
Long-Term DON Content Evaluation of “Norman” vs. “CDC Kendall”
Genotyping
Fungal Cultures
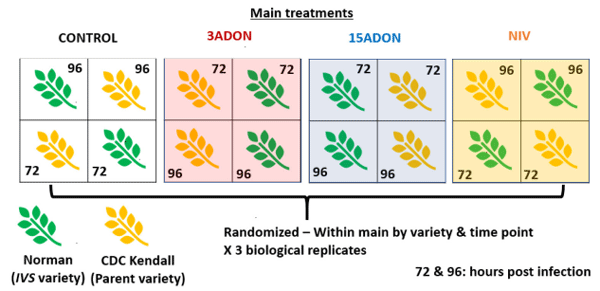
Growth Cabinet—Experimental Design
Statistical Analysis of FHB and Mycotoxin Content
Total RNA Extraction and Quality
RNA-Seq Library Preparation and Sequencing and Quality
Sequence Alignment and Genomic Feature Evaluation
RESULTS
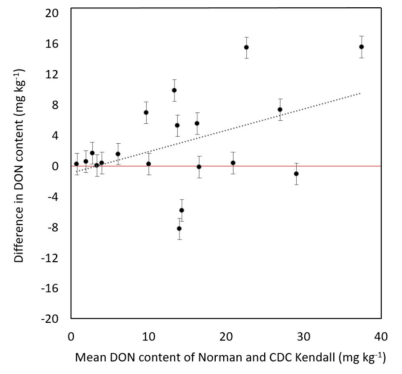
Norman and Its Response to DON Accumulation
Transcriptome Differentiations
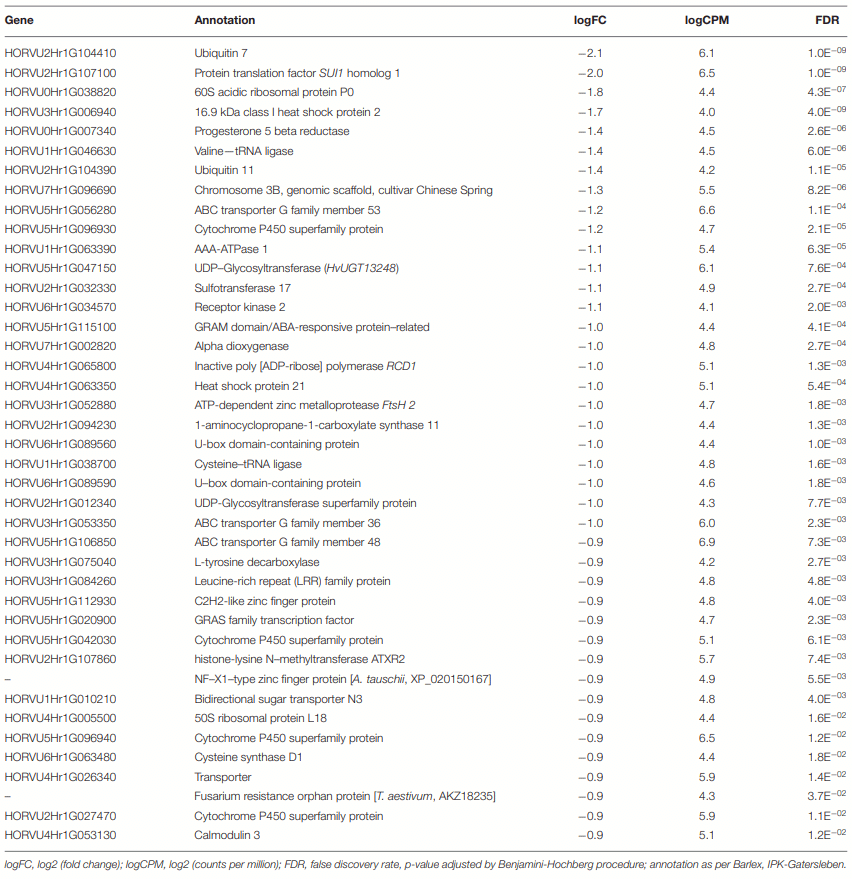
Differential Genes of CDC Kendall
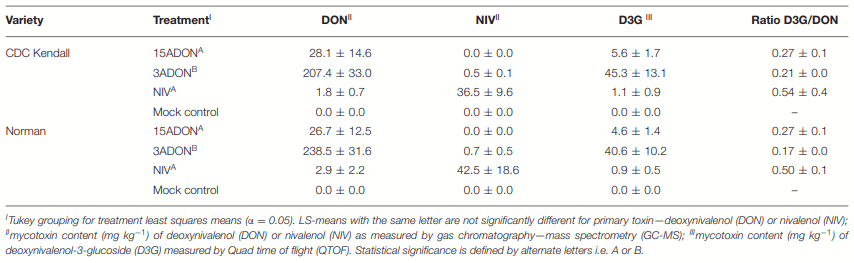
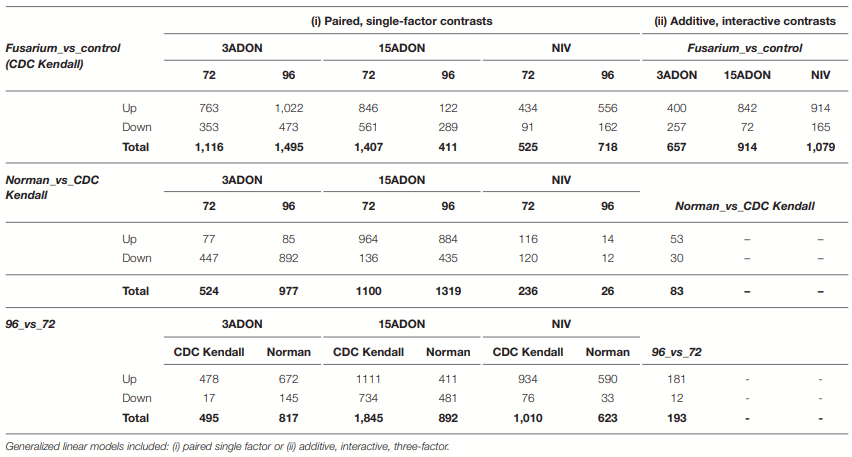
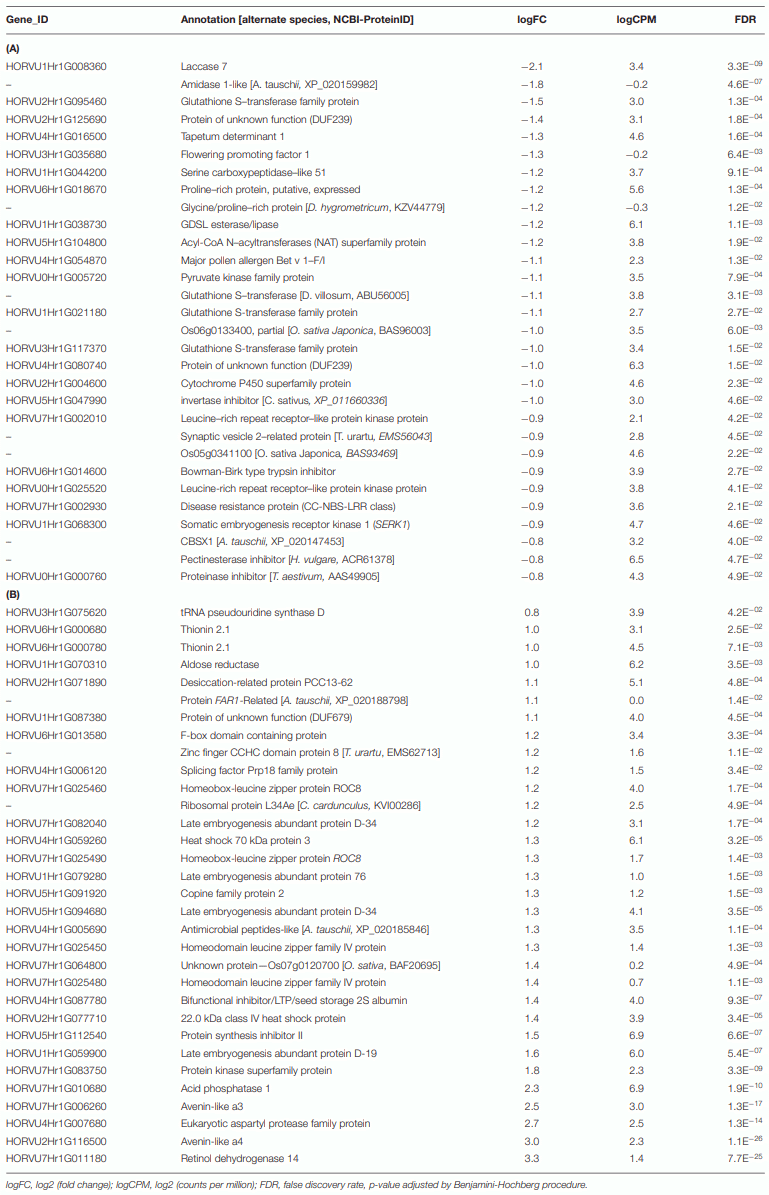
Differential Genes of Norman
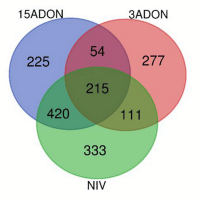
Norman vs. CDC Kendall Control
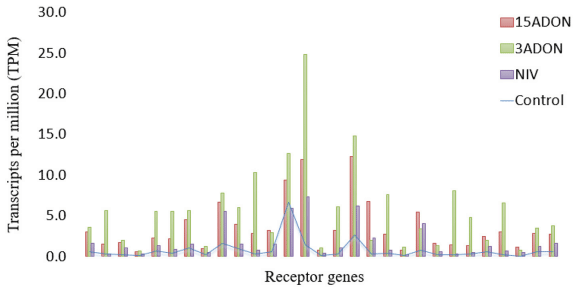
Differential Genes Between Norman and CDC Kendall by Treatment and Time Point
Contrast 72 vs. 96-h Post-infection
Unique Resistance Patterns Were Observed in Norman in 15ADON Treatment at 72 hpi
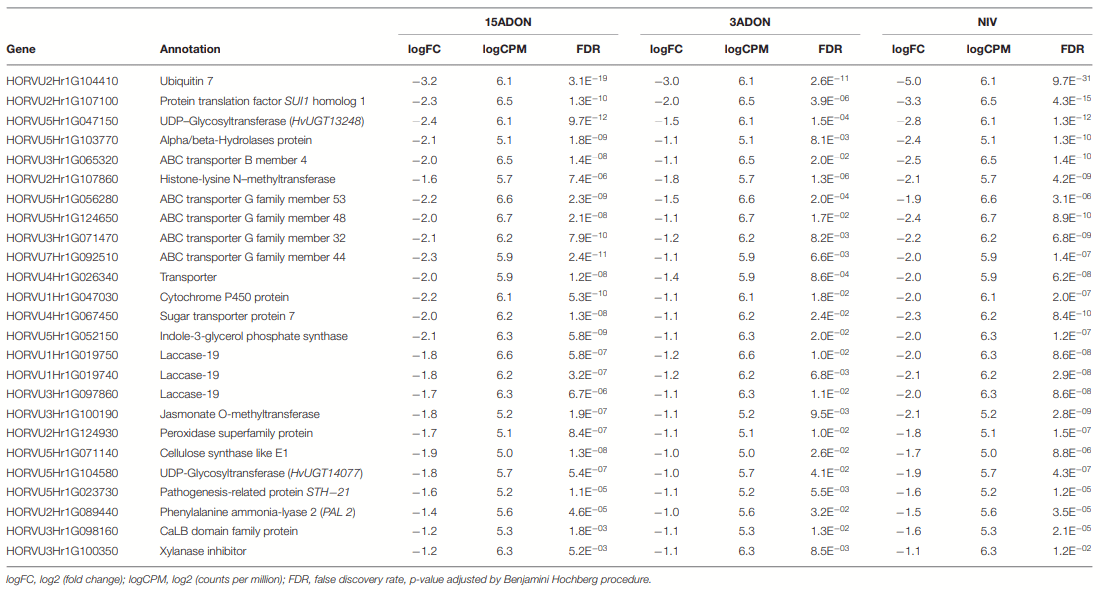
3ADON Treatment at 72 hpi
Norman Displayed Alterations in Responsive Patterns in 15ADON Treatment at 96 hpi
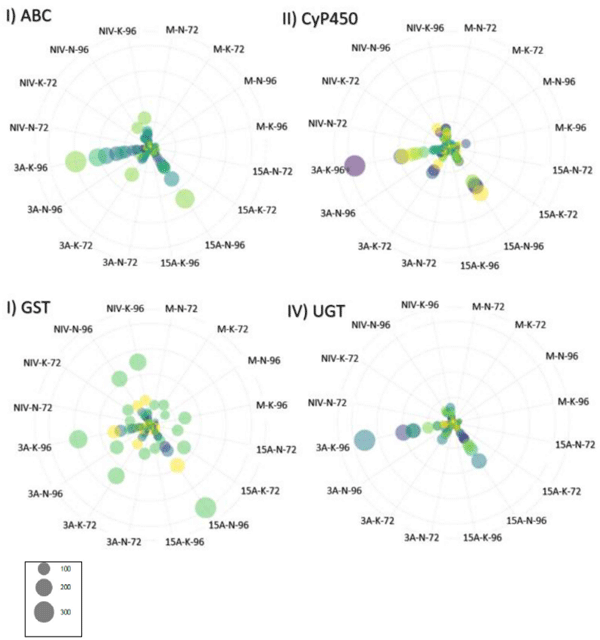
DON Avoidance Mechanisms Were Pronounced in CDC Kendall in 3ADON Treatment at 96 hpi
NIV Treatment at 96 hpi
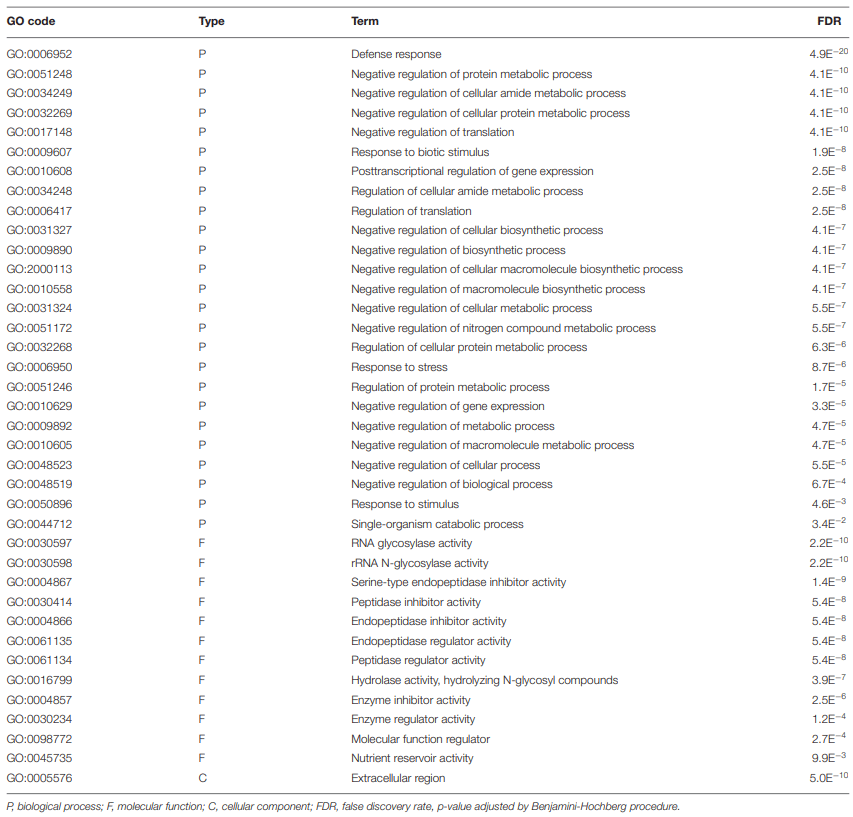
Gene Ontology (GO) Enrichment in Fusarium-Treatment vs. Control
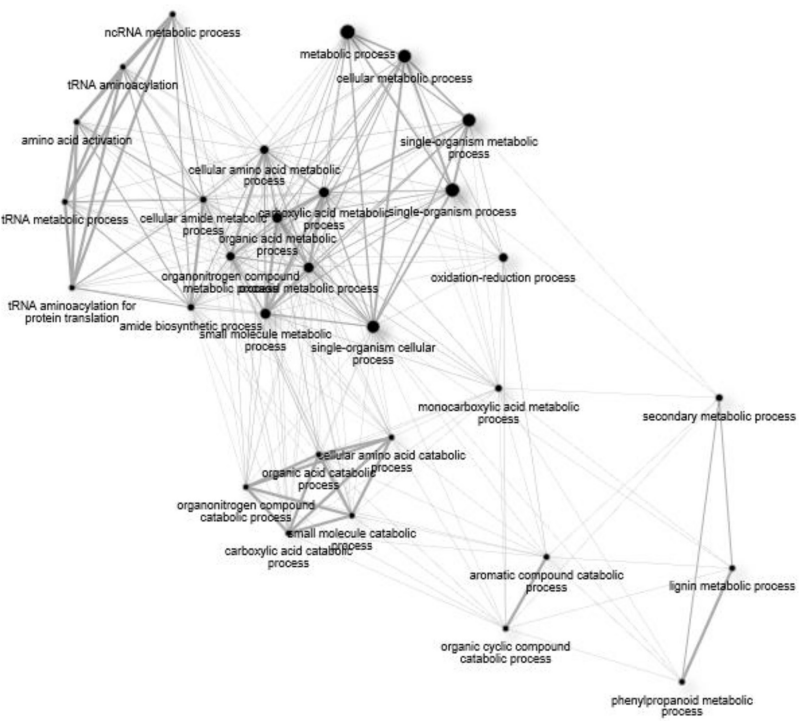
GO Enrichment in 72 vs. 96 hpi
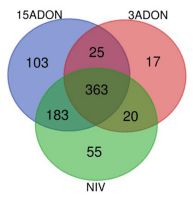
 0.05) Gene Ontology (GO) terms for biological processes common to all treatment groups (15ADON, 3ADON, and NIV)." />
0.05) Gene Ontology (GO) terms for biological processes common to all treatment groups (15ADON, 3ADON, and NIV)." /> GO Enrichment in “Norman” vs. “CDC Kendall”
The Differential Gene Associated With Oxidative Stress Reduction
DISCUSSION
PERMISSION TO REUSE AND COPYRIGHT
DATA AVAILABILITY STATEMENT
AUTHOR CONTRIBUTIONS
FUNDING
ACKNOWLEDGMENTS
SUPPLEMENTARY MATERIAL
Al-Taweel, K., Fernando, W. G. D., and Brûlé-Babel, A. L. (2014). Transcriptome profiling of wheat differentially expressed genes exposed to different chemotypes of Fusarium graminearum. Theor. Appl. Genet. 127, 1703–1718. doi: 10.1007/s00122-014-2333-8
Amarasinghe, C., and Fernando, W. G. D. (2016). Comparative analysis of deoxynivalenol biosynthesis related gene expression among different chemotypes of Fusarium graminearum in spring wheat. Front. Microbiol. 7:1229. doi: 10.3389/fmicb.2016.01229
Amarasinghe, C., Sharanowski, B., and Fernando, W. G. D. (2019). Molecular phylogenetic relationships, trichothecene chemotype diversity and aggressiveness of strains in a global collection of Fusarium graminearum species. Toxins 11:263. doi: 10.3390/toxins11050263
Anand, L., and Rodriguez Lopez, C. M. (2020). chromoMap: an R package for interactive visualization and annotation of chromosomes. bioRxiv 605600. doi: 10.1101/605600
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/ fastqc (accessed March, 2021).
Audenaert, K., Vanheule, A., Höfte, M., and Haesaert, G. (2014). Deoxynivalenol: a major player in the multifaceted response of Fusarium to its environment. Toxins 6, 1–19. doi: 10.3390/toxins6010001
Bai, G., and Shaner, G. (2004). Management and resistance in wheat and barley to fusarium head blight. Annu. Rev. Phytopathol. 42, 135–161. doi: 10.1146/annurev.phyto.42.040803.140340
Bayer, M. M., Rapazote-Flores, P., Ganal, M., Hedley, P. E., Macaulay, M., Plieske, J., et al. (2017). Development and evaluation of a Barley 50k iSelect SNP Array. Front. Plant Sci. 8:1792. doi: 10.3389/fpls.2017.01792
Berthiller, F., Krska, R., Domig, K. J., Kneifel, W., Juge, N., Schuhmacher, R., et al. (2011). Hydrolytic fate of deoxynivalenol-3-glucoside during digestion. Toxicol. Lett. 206, 264–267. doi: 10.1016/j.toxlet.2011.08.006
Boddu, J., Cho, S., Kruger, W. M., and Muehlbauer, G. J. (2006). Transcriptome analysis of the barley-Fusarium graminearum interaction. Mol. Plant Microbe Interact. 19, 407–17. doi: 10.1094/MPMI-19-0407
Boddu, J., Cho, S., and Muehlbauer, G. J. (2007). Transcriptome analysis of trichothecene-induced gene expression in barley. Mol. Plant Microbe Interact. 20, 1364–1375. doi: 10.1094/MPMI-20-11-1364
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boutigny, A.-L., Richard-Forget, F., and Barreau, C. (2008). Natural mechanisms for cereal resistance to the accumulation of Fusarium trichothecenes. Eur. J. Plant Pathol. 121, 411–423. doi: 10.1007/s10658-007-9266-x
Brown, N. A., Antoniw, J., and Hammond-Kosack, K. E. (2012). The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis. PLoS ONE 7:e33731. doi: 10.1371/journal.pone. 0033731
Chamarthi, S. K., Kumar, K., Gunnaiah, R., Ajjamada, K., Dion, Y., and Choo, T. (2014). Identification of fusarium head blight resistance related metabolites specific to doubled-haploid lines in barley. Eur. J. Plant Pathol. 138, 67–78. doi: 10.1007/s10658-013-0302-8
Chawla, H. S., and Wenzel, G. (1987). In-vitro selection for fusaric acid resistant barley plants. Plant Breed. 99, 159–163. doi: 10.1111/j.1439-0523.1987.tb01166.x
Clear, R. M., Tucker, J. R., Gaba, D., Patrick, S. K., Lee, S.-J., Demeke, T., et al. (2013). Deoxynivalenol levels and chemotype frequency in barley varieties inoculated with two chemotypes of Fusarium graminearum. Can J. Plant Pathol. 35, 37–45. doi: 10.1080/07060661.2012.751622
De Bruyne, L., Hofte, M., and De Vleesschauwer, D. (2014). Connecting growth and defense: the emerging roles of brassinosteroids and gibberellins in plant innate immunity. Mol. Plant. 7, 943–959. doi: 10.1093/mp/ssu050
Desjardins, A. E. (2006). “Trichothecenes,” in Fusarium Mycotoxins: Chemistry, Genetics, and Biology, ed A. E. Desjardins (St. Paul, MN: The American Phytopathological Society Press), 13–64.
Desmond, O. J., Manners, J. M., Stephens, A. E., MacLean, D. J., Schenk, P. M., Gardiner, D. M., et al. (2008). The Fusarium mycotoxin deoxynivalenol elicits hydrogen peroxide production, programmed cell death and defence responses in wheat. Mol. Plant Pathol. 9, 435–445. doi: 10.1111/j.1364-3703.2008.00475.x
Dickman, M. B., and Fluhr, R. (2013). Centrality of host cell death in plant-microbe interactions. Annu. Rev. Phytopathol. 51, 543–570. doi: 10.1146/annurev-phyto-081211-173027
Ding, L., Xu, H., Yi, H., Yang, L., Kong, Z., Zhang, L., et al. (2011). Resistance to hemi-biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS ONE 6:e19008. doi: 10.1371/journal.pone.0019008
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Eggert, K., and Pawelzik, E. (2011). Proteome analysis of Fusarium head blight in grains of naked barley (Hordeum vulgare subsp. nudum). Proteomics 11, 972–985. doi: 10.1002/pmic.201000322
Eudes, F., Comeau, A., Rioux, S., and Colin, J. (2008). Trichothecenemediated in vitro selection in wheat for reduced mycotoxin accumulation caused by Fusarium graminearum. Can. J. Plant Sci. 88, 1115–1125. doi: 10.4141/CJPS08060
Fabre, F., Bormann, J., Urbach, S., Roche, S., Langin, T., and Bonhomme, L. (2019). Unbalanced roles of fungal aggressiveness and host cultivars in the establishment of the Fusarium head blight in bread wheat. Front. Microbiol. 10:2857. doi: 10.3389/fmicb.2019.02857
Ferrigo, D., Raiola, A., and Causin, R. (2016). Fusarium toxins in cereals: occurrence, legislation, factors promoting the appearance and their management. Molecules 21:627. doi: 10.3390/molecules21050627
Gardiner, S. A., Boddu, J., Berthiller, F., Hametner, C., Stupar, R. M., Adam, G., et al. (2010). Transcriptome analysis of the barley-deoxynivalenol interaction: evidence for a role of glutathione in deoxynivalenol detoxification. Mol. Plant Microbe Interact. 23, 962–976. doi: 10.1094/MPMI-23-7-0962
Gauthier, L., Atanasova-Penichon, V., Chéreau, S., Richard-Forget, F. (2015). Metabolomics to decipher the chemical defense of cereals against Fusarium graminearum and deoxynivalenol. Int. J. Mol. Sci. 16, 24839–24872. doi: 10.3390/ijms161024839
Ge, S. X., Jung, D., and Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Geddes, J., Eudes, F., Laroche, A., and Selinger, L. B. (2008). Differential expression of proteins in response to the interaction between the pathogen Fusarium graminearum and its host, Hordeumvulgare. Proteomics 8, 545–554. doi: 10.1002/pmic.200700115
Gietler, M., Fidler, J., Labudda, M., and Nykiel, M. (2020). Abscisic acid-enemy or savior in the response of cereals to abiotic and biotic stresses? Int. J. Mol. Sci. 21:4607. doi: 10.3390/ijms21134607
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–222. doi: 10.1146/annurev.phyto.43.040204.135923
Gottwald, S., Samans, B., Luck, S., and Friedt, W. (2012). Jasmonate and ethylene dependent defence gene expression and suppression of fungal virulence factors: two essential mechanisms of Fusarium head blight resistance in wheat? BMC Genomics 13:369. doi: 10.1186/1471-2164-13-369
Güldener, U., Seong, K. Y., Boddu, J., Cho, S., Trail, F., Xu, J. R., et al. (2006). Development of a Fusarium graminearum Affymetrix GeneChip for profiling fungal gene expression in vitro and in planta. Fungal Genet. Biol. 43, 316–325. doi: 10.1016/j.fgb.2006.01.005
Habler, K., Geissinger, C., Hofer, K., Schüler, J., Moghari, S., Hess, M., et al. (2017). Fate of Fusarium toxins during brewing. J. Agric. Food Chem. 65, 190–198. doi: 10.1021/acs.jafc.6b04182
Hao, G., Bakker, M. G., and Kim, H.-S. (2020). Enhanced resistance to Fusarium graminearum in transgenic Arabidopsis plants expressing a modified plant thionin. Phytopathology 110, 1956–1066. doi: 10.1094/PHYTO-12-19-0447-R
Hao, Q., Wang, W., Han, X., Wu, J., Lyu, B., Chen, F., et al. (2018). Isochorismate-based salicylic acid biosynthesis confers basal resistance to Fusarium graminearum in barley. Mol. Plant Pathol. 19, 1995–2010. doi: 10.1111/mpp.12675
Huang, S., Zhang, X., and Fernando, W. G. D. (2020). Directing trophic divergence in plant-pathogen interactions: antagonistic phytohormones with NO doubt? Front. Plant Sci. 11:1850. doi: 10.3389/fpls.2020.600063
Huang, Y., Li, L., Smith, K. P., and Muehlbauer, G. J. (2016). Differential transcriptomic responses to Fusarium graminearum infection in two barley quantitative trait loci associated with Fusarium head blight resistance. BMC Genom. 17:387. doi: 10.1186/s12864-016-2716-0
Jansen, C., von Wettstein, D., Schäfer, W., Kogel, K.-H., Felk, A., and Maier, F. J. (2005). Infection patterns in barley and wheat spikes inoculated with wild-type and trichodiene synthase gene disrupted Fusarium graminearum. Proc. Natl. Acad. Sci. U.S.A. 102, 16892–16897. doi: 10.1073/pnas.0508467102
Kelly, A. C., and Ward, T. J. (2018). Population genomics of Fusarium graminearum reveals signatures of divergent evolution within a major cereal pathogen. PLoS ONE 13:e0194616. doi: 10.1371/journal.pone.0194616
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360. doi: 10.1038/nmeth.3317
Kluger, B., Bueschl, C., Lemmens, M., Michlmayr, H., Malachova, A., Koutnik, A., et al. (2015). Biotransformation of the mycotoxin deoxynivalenol in fusarium resistant and susceptible near isogenic wheat lines. PLoS ONE 10:e0119656. doi: 10.1371/journal.pone.0119656
Koubaa, S., and Brini, F. (2020). Functional analysis of a wheat group 3 late embryogenesis abundant protein (TdLEA3) in Arabidopsis thaliana under abiotic and biotic stresses. Plant Physiol. Biochem. 156, 396–406. doi: 10.1016/j.plaphy.2020.09.028
Kumaraswamy, G. K., Kushalappa, A. C., Choo, T. M., Dion, Y., and Rioux, S. (2012). Differential metabolic response of barley genotypes, varying in resistance, to trichothecene-producing and -nonproducing (tri5-) isolates of Fusarium graminearum. Plant Pathol. 61, 509–521. doi: 10.1111/j.1365-3059.2011.02528.x
Lamb, C., and Dixon, R. A. (1997). The oxidative burst in plant disease resistance. Annu. Rev. Plant Physiol. 48, 251–275. doi: 10.1146/annurev.arplant.48.1.251
Legge, W. G., Tucker, J. R., Bizimungu, B., Tekauz, A., Noll, J. S., Fetch, J.r,., et al. (2011). Norman barley. Can. J. Plant Sci. 91, 1105–1113. doi: 10.4141/cjps2010-020
Li, G., and Yen, Y. (2008). Jasmonate and ethylene signaling pathway may mediate Fusarium head blight resistance in wheat. Crop Sci. 48, 1888–1896. doi: 10.2135/cropsci2008.02.0097
Li, X., Shin, S., Heinen, S., Dill-Macky, R., Berthiller, F., Nersesian, N., et al. (2015). Transgenic wheat expressing a barley UDP-glucosyltransferase detoxifies deoxynivalenol and provides high levels of resistance to Fusarium graminearum. Mol. Plant Microbe Interact. 28, 1237–1246. doi: 10.1094/MPMI-03-15-0062-R
Liu, J., Li, L., Foroud, N. A., Gong, X., Li, C., and Li, T. (2019). Proteomics of bulked rachides combined with documented QTL uncovers genotype nonspecific players of the Fusarium head blight responses in wheat. Phytopathology 1, 111–119. doi: 10.1094/PHYTO-03-18-0086-R
López, M. A., Bannenberg, G., and Castresana, C. (2008). Controlling hormone signaling is a plant and pathogen challenge for growth and survival. Curr. Opin. Plant Biol. 11, 420–427. doi: 10.1016/j.pbi.2008.05.002
Makandar, R., Nalam, V. J., Lee, H., Trick, H. N., Dong, Y., and Shah, J. (2012). Salicylic acid regulates basal resistance to Fusarium head blight in wheat. Mol. Plant Microbe Interact. 25, 431–439. doi: 10.1094/MPMI-09-11-0232
Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S. O., Wicker, T., et al. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433. dx. doi: 10.1038/nature22043
Miller, J. D., Greenhalgh, R., Wang, Y., and Lu, M. (1991). Trichothecene chemotypes of three Fusarium species. Mycologia 83, 121–130. doi: 10.1080/00275514.1991.12025988
Murase, K., Hirano, Y., Sun, T. P., and Hakoshima, T. (2008). Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 456, 459–463. doi: 10.1038/nature07519
Pekkarinen, A. I., Longstaff, C., and Jones, B. L. (2007). Kinetics of the inhibition of Fusarium serine proteinases by barley (Hordeum vulgare L.) inhibitors. J. Agric. Food Chem. 55, 2736–2742. doi: 10.1021/jf0631777
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33:2. doi: 10.1038/nbt.3122
Peters, J., van Dam, R., van Doorn, R., Katerere, D., Berthiller, F., Haasnoot, W., et al. (2017). Mycotoxin profiling of 1000 beer samples with a special focus on craft beer. PLoS ONE 12:e0185887. doi: 10.1371/journal.pone.0185887
Petti, C., Khan, M., and Doohan, F. (2010). Lipid transfer proteins and protease inhibitors as key factors in the priming of barley responses to Fusarium head blight disease by a biocontrol strain of Pseudomonas fluorescens. Funct. Integr. Genomics. 10, 619–627. doi: 10.1007/s10142-010-0177-0
Poland, J. A., Balint-Kurti, P. J., Wisser, R. J., Pratt, R. C., and Nelson, R. J. (2009). Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14, 21–29. doi: 10.1016/j.tplants.2008.10.006
Ponts, N., Couedelo, L., Pinson-Gadais, L., Verdal-Bonnin, M. N., Barreau, C., and Richard-Forget, F. (2009). Fusarium response to oxidative stress by H2O2 is trichothecene chemotype-dependent. FEMS Microbiol. Lett. 293, 255–262. doi: 10.1111/j.1574-6968.2009.01521.x
Ponts, N., Pinson-Gadais, L., Barreau, C., Richard-Forget, F., and Ouellet, T. (2007). Exogenous H2O2 and catalase treatments interfere with Tri genes expression in liquid cultures of Fusarium graminearum. FEBS Lett. 581, 443–447. doi: 10.1016/j.febslet.2007.01.003
Ponts, N., Pinson-Gadais, L., Boutigny, A.-L., Barreau, C., and Richard-Forget, F. (2011). Cinnamic derived acids significantly affect Fusarium graminearum growth and in vitro synthesis of type B trichothecenes. Phytopathology 101, 929–934. doi: 10.1094/PHYTO-09-10-0230
Puri, K. D., Yan, C., Leng, Y., and Zhong, S. (2016). RNA-Seq revealed differences in transcriptomes between 3ADON and 15ADON populations of Fusarium graminearum in vitro and In Planta. PLoS ONE 11:e0163803. doi: 10.1371/journal.pone.0163803
Puri, K. D., and Zhong, S. (2010). The 3ADON population of Fusarium graminearum found in North Dakota is more aggressive and produces a higher level of DON than the prevalent 15ADON population in spring wheat. Phytopathology 100, 1007–1014. doi: 10.1094/PHYTO-12-09-0332
Qi, P.-F., Balcerzak, M., Rocheleau, H., Leung, W., Wei, Y.-M., Zheng, Y.-L., et al. (2016). Jasmonic acid and abscisic acid play important roles in hostpathogen interaction between Fusarium graminearum and wheat during the early stages of fusarium head blight. Physiol. Mol. Plant Pathol. 93, 39–48. doi: 10.1016/j.pmpp,.2015.12.004
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: https:// www.R-project.org/ (accessed July 22, 2021).
Rao, S., and Sandhya, H. (2016). “in vitro selection of disease-resistant plants,” in Plant Tissue Culture: Propagation, Conservation and Crop Improvement, eds M. Anis M and N. Ahmad (Singapore: Springer), 395–417. doi: 10.1007/978-981-10-1917-3_17
Rasmussen, S. K., Welinder, K. G., and Hejgaard, J. (1991). cDNA cloning, characterization and expression of an endosperm-specific barley peroxidase. Plant Mol. Biol. 16, 317–327. doi: 10.1007/B. F.00020562
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/ btp616 SAS Institute Inc (1989-2019). SAS JMP R 13.2.1. Cary, NC: SAS Institute Inc.
SAS R 9.4, SAS Institute Inc (2013). Statements: Reference. Cary, NC: SAS R 9.4, SAS Institute Inc.
Saville, R. J., Gosman, N., Burt, C. J., Makepeace, J., Steed, A., Corbitt, M., et al. (2012). The ‘Green Revolution’ dwarfing genes play a role in disease resistance in Triticum aestivum and Hordeum vulgare. J. Exp. Bot. 63, 1271–1283. doi: 10.1093/jxb/err350
Schweiger, W., Boddu, J., Shin, S., Poppenberger, B., Berthiller, F., Lemmens, M., et al. (2010). Validation of a candidate deoxynivalenol-inactivating UDPglucosyltransferase from barley by heterologous expression in yeast. Mol. Plant Microbe Interact. 23, 977–986. doi: 10.1094/MPMI-23-7-0977
Sinha, R. C., and Savard, M. E. (1996). Comparison of immunoassay and gas chromatography methods for the detection of the mycotoxin deoxynivalenol in grain samples. Can. J. Plant Pathol. 18, 233–236. doi: 10.1080/07060669609500617
Taheri, P. (2018). Cereal diseases caused by Fusarium graminearum: from biology of the pathogen to oxidative burst-related host defense responses. Eur. J. Plant Pathol. 152, 1–20. doi: 10.1007/s10658-018-1471-2
Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z., et al. (2017). agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45:gkx382. doi: 10.1093/nar/gkx382
Trail, F. (2009). For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 149, 103–110. doi: 10.1104/pp.108.129684
Tucker, J. R., Badea, A., Blagden, R., Pleskach, K., Tittlemier, S. A., and Fernando, W. G. D. (2019). Deoxynivalenol-3-glucoside content is highly associated with deoxynivalenol levels in two-row barley genotypes of importance to Canadian barley breeding programs. Toxins 11:319. doi: 10.3390/toxins11060319 von der
Ohe, C., Gauthier, V., Tamburic-Ilincic, L., Brule-Babel, A., Fernando, W. G. D., Clear, R., et al. (2010). A comparison of aggressiveness and deoxynivalenol production between Canadian Fusarium graminearum isolates with 3-acetyl and 15-acetyldeoxynivalenol chemotypes in field-grown spring wheat. Eur. J. Plant Pathol. 127, 407–417. doi: 10.1007/s10658-010-9607-z
Walkowiak, S., Bonner, C. T., Wang, L., Blackwell, B., Rowland, O., and Subramaniam, R. (2015). Intraspecies interaction of Fusarium graminearum contributes to reduced toxin production and virulence. Mol. Plant Microbe Interact. 28, 1256–1267. doi: 10.1094/MPMI-06-15-0120-R
Wang, L., Li, Q., Liu, Z., Surendra, A., Pan, Y., Yifeng Li, L., et al. (2018). Integrated transcriptome and hormone profiling highlight the role of multiple phytohormone pathways in wheat resistance against fusarium head blight. PLoS ONE 13:e0207036. doi: 10.1371/journal.pone.0207036
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Ward, T. J., Clear, R. M., Rooney, A. P., O’Donnell, K., Gaba, D., Patrick, S., et al. (2008). An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet. Biol. 45, 473–484. doi: 10.1016/j.fgb.2007.10.003
Wilson, W. B., Dahl, B., and Nganje, W. (2018). Economic costs of Fusarium Head Blight, scab and deoxynivalenol. World Mycotoxin J. 11, 291–302. doi: 10.3920/WMJ2017.2204
Wu, J., Wang, Y., Kim, S. T., Kim, S. G., and Kang, K. Y. (2013). Characterization of a newly identified rice chitinase-like protein (OsCLP) homologous to xylanase inhibitor. BMC Biotechnol. 13:4. doi: 10.1186/1472-6750-13-4
Xing, L.-P., He, L.-Q., Xiao, J., Chen, Q.-G., Li, M.-H., Shang, Y., et al. (2017). An UDP-Glucosyltransferase gene from barley confers disease resistance to Fusarium head blight. Plant Mol. Biol. Rep. 35, 224–236. doi: 10.1007/s11105-016-1014-y
Zantinge, J., Kumar, K., Xi, K., Johns, M., Murray, A., Jones, T., et al. (2010). Comparison of barley seed proteomic profiles associated with Fusarium head blight reaction. Can. J. Plant Pathol. 32, 496–512. doi: 10.1080/07060661.2010.529252
Zhou, B., Schwarz, P., He, G.-Q., Gillespie, J., and Horsley, R. (2008). Effect of enzyme pretreatments on the determination of deoxynivalenol in barley. J. Am. Soc. Brew. Chem. 66, 103–108. doi: 10.1094/ASBCJ-2008-0226-01







.jpg&w=3840&q=75)