Toxic effects of mycotoxins: importance of biotransformation systems
The fate of a mycotoxin in the animal body depends upon the extent and rate of its absorption from the gastrointestinal tract, its distribution, its binding or localization in tissues, its biotransformation and its excretion processes. The rate of each of these events, which contribute to both toxicokinetics and toxicodynamics of a toxin, is determined by the chemical and physical properties of the compound and by its interaction with tissues responsible for elimination.
The enzymatic biotransformation changes undergone by a toxin in the body usually result in the attenuation or loss of toxicological activity. The term ‘detoxification’ describes the result of such metabolic changes. However, this is not the only possibility: a toxicologically active metabolite may be formed from a less active parent toxin, which would lead to bioactivation of such compounds.
The main organ concerned with drug metabolism is the liver, and many toxins are substrates for the microsomal enzyme systems of hepatocytes. Nevertheless, kidney, lung, intestinal mucosa or nervous tissues also contain important xenobiotic-metabolizing enzymes. In the case of breeding animals, of special concern are the catalytic activities developed by ruminal or intestinal microflora, since mycotoxins enter the organism by the oral route, as do all food or feed contaminants.
The aim of the present paper is to present a rapid overview of mycotoxin toxicity, to review characteristics of biotransformation pathways undergone by the major toxins in animals or humans and to describe consequences of these metabolic pathways in terms of toxicity and particularly of bioactivation or detoxification processes.
General overview of mycotoxin toxicity
Based on their diverse chemical structure, mycotoxins may cause a wide range of toxic responses ranging from acute lethality to subclinical biochemical alterations. Regarding acute toxicity, these toxins can be divided into three groups: (i) mycotoxins of low toxicity (oral LD50 lower than 100 mg/kg body weight in mice) including fumonisins, zearalenone, citrinin, penicillic acid, mycophenolic acid, sterigmatocystin, sporidesmin, tenuazonic acid, rubratoxins, (ii) mycotoxins of intermediate toxicity (oral LD50 between 20 and 100 mg/kg body weight in mice) including deoxynivalenol, ochratoxin A, patulin, gliotoxin, cyclopiazonic acid, verruculogen, and (iii) mycotoxins of high toxicity (oral LD50 lower than 10 mg/kg body weight in mice) including penitrem, aflatoxin B1, T-2 toxin, diacetoxyscirpenol and fusarenon X. In fact, animals demonstrate varying sensitivities to the mycotoxins, which may be based on species, sex, breed, strain, physiology, age, nutrition, other diseases, presence of other environmental factors and management. This is particularly true for aflatoxins for which ducklings, cats, pigs and rabbits would be the more sensitive animal species. Generally, for most mycotoxins, if high doses result in acute mycotoxicoses and may be lethal, sublethal doses would induce mycotoxicoses of varying clinical signs and symptoms. These signs could include decreased feed conversion efficiency (trichothecenes), delayed growth of litters (aflatoxins), oestrogenic effects (zearalenone), embryonic mortality or teratogenicity (aflatoxins, ochratoxin A, T-2 toxin) (Hayes, 1973).
For some mycotoxins at low doses, the effects on animals or humans may be subtle and would affect only the immune system (Oswald and Comera, 1998), so the only visible signs correspond to those of secondary infections to which the toxin predisposed the consumer. On the other hand, the possible long-term effects of mycotoxins as mutagens and carcinogens have attracted the attention of scientists. By considering all experimental and epidemiological data, the International Agency for Research on Cancer (IARC) initiated a program for the evaluation of the carcinogenic risk to humans of chemicals (Castegnaro and MacGregor, 1998). Many mycotoxins have been evaluated in this system. In this evaluation, aflatoxin B1 has been recognized as carcinogenic in humans (group 1), whereas aflatoxin M1, griseofulvin, sterigmatocystin, ochratoxin A and recently fumonisin B1, have been classified as possible carcinogens with evidence in animals (group 2B). Both patulin, citrinin, penicillic acid and certain fusariotoxins were not classifiable in terms of carcinogenicity for humans.
Toxic effects in relation to mycotoxin biotransformations
Since mycotoxins are natural products, there is a lack in knowledge of metabolic pathways regarding many of the less investigated toxins. By contrast, the biological fate of certain toxins like aflatoxin B1, ochratoxin A, trichothecenes, zearalenone or fumonisin B1 have been described.
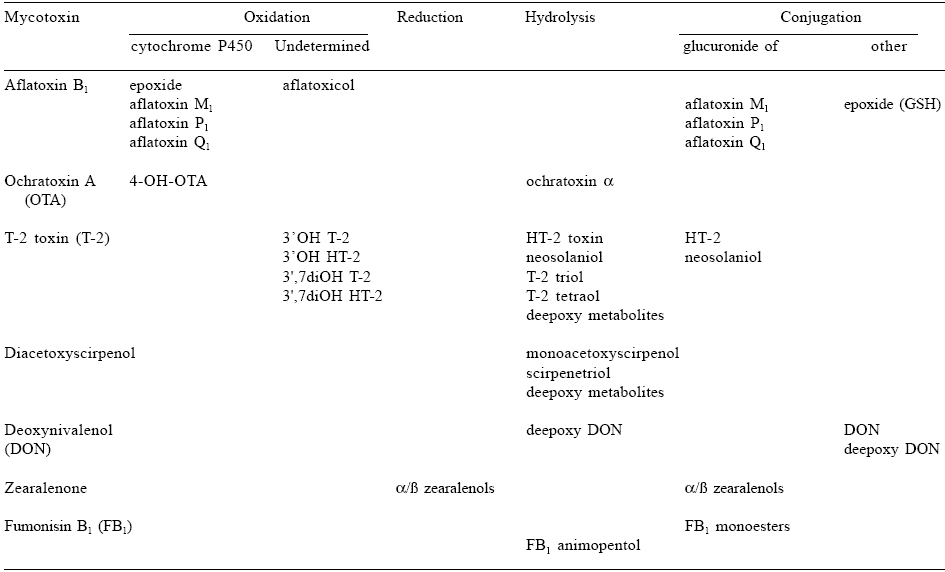
The reactions of xenobiotic metabolism have been divided into two phases. Phase I reactions are those in which there is a change in the parent molecule, for example oxidation, reduction or hydrolysis. Microsomal cytochromes P450, flavin-containing monooxygenases, prostaglandin synthases, amine oxydases and alcohol dehydrogenases are the major enzymes involved in oxidations, whereas reductive metabolism is mainly governed by epoxide hydrolases, aldehyde or ketone reductases. Furthermore, mammalian tissues and body fluids contain a large number of nonspecific esterases or amidases that can hydrolyse ester or amide linkages in xenobiotics. Phase II reactions are those in which a conjugate is formed with a toxin or a toxin metabolite produced in a phase I reaction. The major conjugating enzymes are known to be microsomal glucuronosyltransferases and cytosolic sulfotransferases, methyltransferases, N-acetyltransferases, glutathione S-transferases and aminoacyltransferases. Data obtained in the last two decades have given a better understanding of biotransformation pathways of mycotoxins and their consequences in terms of metabolites occurring in animal-derived food products. The major biological enzyme systems involved in these bioconversions are listed in Table 1.
Table 1. Major biotransformation pathways undergone by mycotoxins in biological systems.
To enlarge the image, click here
From these data, it is clear that certain enzymes like cytochromes P450 are able to bioactivate mycotoxins in deleterious intermediates. In other cases, reductases or hydrolases would biotransform parent toxins into toxicologically active metabolites such as zearalenols or HT-2 toxin. By contrast, conjugations to glucuronic acid, sulfuric acid or glutathione can be considered as detoxifying enzymes leading to conjugates less toxic than the parent mycotoxin. The following five sections address mycotoxins of human health concern from exposure to animalderived products. There are many additional mycotoxicoses that can affect animal health and economics (Le Bars and Le Bars, 1996). The toxins addressed in this review are aflatoxin B1, ochratoxin A, trichothecenes, zearalenone and fumonisin B1. Each mycotoxin is discussed separately, with description of its biological metabolism considered as a potential determinant of its toxic effects.
AFLATOXINS
In the case of aflatoxins, biotransformation plays a major role in the disposition and toxicological activity of these toxins. Bioactivation has been demonstrated as a prerequisite in most of the toxic and carcinogenic effects of aflatoxins. These aspects have been recently detailed in a comprehensive review (Eaton and Groopman, 1994). Consequently, much attention has focused on the biotransformation of aflatoxin B1 (AFB1), the most naturally occurring and toxic compound of this group of toxins produced by Aspergillus flavus and A. parasiticus. The metabolism of AFB1 would involve the four following reactions: reduction, hydroxylation, epoxidation and O-dealkylation.
The ketone reduction of AFB1 into aflatoxicol would be associated with NADPH reductase activity while the other reactions are primarily carried out by the cytochrome P450 enzyme superfamily (P450, CYP). This conclusion is based on the requirement for NADPH in in vitro metabolism experiments, the alteration of microsomal activation in the presence of P450 inducers or inhibitors, the successful activation of AFB1 in in vitro reconstituted systems including purified or engineeredexpressed P450 and the specific inhibition exerted by antibodies against certain P450 isoenzymes (Essigman et al., 1982; Aoyama et al., 1990, Shimada and Guengerich, 1989; Crespi et al., 1991).
Epoxidation of AFB1 into a 8,9 exo-epoxide would be at the origin of the mutagenicity and carcinogenicity of this mycotoxin. Even though this epoxide has not been isolated due to its reactivity, the major DNA adduct formed in vivo or in vitro is 8,9 dihydro 8-(N7guanyl)-9 hydroxyAFB1 (Essigmann et al., 1982; Harrison and Gamer, 1991; Ball et al., 1990) whereas the extent of in vivo covalent binding of radiolabelled AFB1 to DNA in hepatic systems has been correlated with the incidence of cancer (Essigmann et al. 1982; Ball et al., 1990; Lutz et al., 1980).
In addition to epoxidation, P450-dependent monooxygenases would oxidize AFB1 to hydroxylated or dealkylated products including aflatoxins M1 (AFM1), Q1 (AFQ1), B2a (AFB2a) or P1 (AFB1), respectively. In all cases, efforts have been made to identify which cytochrome P450 isoforms are specifically involved in these various oxidations. Recently, by using human microsomal or complementary DNA-expressed cytochrome P450, Gallagher et al. (1994) demonstrated that P4501A2 is the principal human P450 enzyme responsible for the activation of AFB1 to its 8,9 epoxide at substrate levels more reflective of dietary exposure, since P4503A4 would contribute to epoxidation at relatively high substrate concentrations. On the other hand, P4501A2 would be involved in the oxidation to AFM1 and P4503A4 responsible for the conversion of AFB1 to AFQ1 in human liver microsomes. Furthermore, if the role of P4502 family enzymes remains undetermined or unclear, the human polymorphic P4502D6 (debrisoquine hydroxylase activity) does not appear to be implicated significantly in aflatoxin metabolism (Forrester et al., 1990). Additional data have revealed AFB1 activation via lipid hydroperoxide-dependent mechanisms. Therefore, the formation of the electrophilic DNA binding epoxide would also be catalysed by microsomal prostaglandin H synthase (Battista and Mamett, 1985; Liu et al., 1990) and by cytosolic lipoxygenases (Liu and Massey, 1992).
Even though AFB1 activation to epoxide prior to DNA binding and carcinogenesis is well established, levels of activating enzymes are not the unique determinants of sensitivity to AFB1 toxicity. The activity of detoxifying enzymes such as glutathione (GST) or glucuronyl transferases must also be considered.
In this respect, GST-catalysed conjugation of activated AFB1 is thought to be the most important detoxification system and to play a key role in the protection of tissues from AFB1 toxicity (Neal and Green, 1983). Using various methodological approaches, it was demonstrated that both α- and μ classes of GST were able to conjugate AFB1-8,9 epoxide (Coles et al., 1985; Gopalan et al., 1992) as a function of species differences in microsomal activation resulting in different ratios of exo- and endo-epoxides. In humans, Liu et al. (1991) demonstrated that incubation of liver preparations from patients expressing high hepatic GSTM1 activity inhibited the formation of AFB1-DNA adducts more than those from individuals with lower corresponding activity.
The hydroxylated or O-dealkylated metabolites like AFM1, AFP1, or AFQ1 are readily conjugated to glucuronic acid or sulfate and furtherly excreted in bile or urine of several species including trout, mouse, hamster, rabbit and monkey (Ball and Coulombe, 1991; Wei et al., 1985; Valsta et al., 1988; Raj and Lotlikar, 1984). However, the toxicological significance of glucuronidation and sulfation would be even more likely minimal because hydroxylated AFB1 metabolites have low toxicity.
Actually, the carcinogenic potency of the parent AFB1 is 15 times that of AFM1 in the rat (Campbell and Hayes, 1976) and over 100 times that of AFQ1 in trout (Coulombe et al., 1982). Concerning the microbial metabolism of AFB1, no metabolic effect by intact rumen fluid, rumen protozoa or rumen bacteria on AFB1 was demonstrated, in contrast to other mycotoxins (Kiessling et al., 1984).
OCHRATOXIN A
Ochratoxin A (OTA) undergoes rapid biotransformation to ochratoxin α (OTα) (Nel and Purchase, 1968). This compound was subsquently identified as the dihydro-isocoumarin derivative produced by hydrolysis of the peptide bond of OTA and has since been recovered in all metabolic studies of OTA in rats, regardless of the route of administration (Van Walbeek et al., 1971; Galtier, 1974; Galtier et al., 1979).
Because of the importance of this derivative in all animals tested, several studies were designed to determine the origin of the biotransformation. Pitout (1969) concluded that the mycotoxin could be hydrolysed by carboxypeptidase A and possibly by trypsin, α-chymotrypsin and cathespin C. Accordingly, OTα was found only in the large intestine of orally treated rats (Galtier et al., 1979) whereas hydrolysis by the bacterial flora of rat caecum was demonstrated after incubation of the native toxin in rat caecal contents. Similar results were obtained with ruminal fluid from cows and sheep, which in some cases esterified OTA to ochratoxin C (Galtier and Alvinerie, 1976). After centrifugation of ruminal fluid, only a fraction containing protozoa could hydrolyse the mycotoxin efficiently, whereas the sterile supernatant, like that found in caecal contents, neither transformed nor destroyed OTA.
Subsequently, Kiessling et al. (1984) demonstrated a decreased capacity of ruminal fluid to cleave OTA to OTα and phenylalanine after feeding, although this activity was gradually restored by the next feeding time.
Intraperitoneal injection of OTA to rats resulted in urinary excretion of a compound corresponding to 4-hydroxyochratoxin-A (4OH-OTA) (Hutchinson and Steyn, 1971). This metabolite was the major one obtained after incubating OTA with rat liver microsomes and NADPH whereas this metabolism was inhibited by carbon monoxide and metyrapone.
The rate of formation increased in microsomal preparations from phenobarbital-treated animals, and a type-I spectrum appeared after binding of the toxin to microsomes. These findings suggest the involvement of cytochrome P450 in the 4-hydroxylation of OTA by hepatic microsomes (Stormer and Pedersen, 1980). When OTA was incubated with pig liver microsomes, two epimeric hydroxylated metabolites (4R)- and (4S)-OH-OTA were formed in approximately equal amounts. These metabolites were also formed, but in a different ratio, in experiments with microsomes from human and rat liver (Stormer et al., 1981). The formation of another metabolite, identified as 10-OH-OTA has been described after incubation of ochratoxin A with rabbit liver microsomes, although this compound has not yet been described in vivo (Stormer et al., 1983).
Possible polymorphism in 4-hydroxylation of the mycotoxin was investigated and gave further support to a possible co-segregation of the genes that regulate OTA and debrisoquine 4-hydroxylation (Castegnaro et al., 1989).
Regarding the toxicity of OTA metabolites, it has been established that the toxicity of the hydrolysate OTα was very low whereas ochratoxin C was as toxic as OTA (Chu et al., 1972). When [4R]-OHOTA and OTα were investigated for their immunosuppresive properties in mice, only the 4- OH-OTA was almost as effective an immunosuppressor as OTA (Creppy et al., 1983).
Since OTA could be associated with an endemic nephropathy in the Balkans, mutagenic properties of this mycotoxin have been investigated. In this respect, it was demonstrated that the mutation frequency of OTA was increased in cell lines stably expressing human P450 1A1, 1A2, 2C10 or 3A4. By contrast, neither in vector-infected cells nor in P450 2D6- or 2E1-expressing cells was an increase of mutation frequency observed (De Groene et al., 1996). In consequence, like AFB1, the mutagenicity of OTA could be related to its previous bioactivation through the oxidizing activity of certain liver microsomal cytochromes P450 and particularly P4501A and 3A subfamilies.
TRICHOTHECENES
A comprehensive review on the metabolism of T-2 toxin (T-2) has been published (Yagen and Bialer, 1993). Briefly, this mycotoxin is known to be extensively metabolized. For instance, using isolated perfused rat liver, Gareis et al. (1986) showed that T-2 was metabolized in two phases. In the first step, it was mainly deacetylated at the C-4 position producing HT-2 toxin (HT-2), and was to a small degree deacetylated at the C-15 position giving T-2 triol. In a second step, these metabolites were conjugated with glucuronic acid and excreted in bile. Two other hydroxy metabolites were also produced in small amounts: 3’OH-HT-2 and 3',7diOH-HT-2. These metabolic pathways were confirmed in vivo using various animal species (Yagen and Bialer, 1993).
In additional studies using other in vitro models like red blood cells (Johnsen et al., 1988), ester hydrolysis at the C-8 position produced neosolaniol and 4-deacetylneosolaniol, which was further hydrolysed to T-2 tetraol. Therefore a large variety of investigations has demonstrated the extreme complexity of T-2 metabolism in biological matrices. In breeding animals, Bauer (1995) identified HT-2, 3’OH-T-2, 3’OH-HT-2, neosolaniol, 4-deacetylneosolaniol, T-2triol, T-2tetraol and deepoxyT- 2tetraol as the major metabolites of T-2 in pigs. Beside unknown metabolites, a similar pattern of metabolites including unmetabolized T-2, HT-2, neosolaniol and 4-deacetylneosolaniol was obtained in cows (Yoschizawa et al., 1981). Regarding the fate of T-2 in microbial flora, the ability of the protozoa and bacteria in intact rumen fluid to metabolize T-2 was first demonstrated by Kiessling et al. (1984). Rat intestinal microflora metabolized T-2 to deepoxyT-2 and deepoxyT-2triol (Swanson et al., 1988). Concerning the toxicological significance of T-2 metabolites, it is now well established that HT- 2 toxicity is similar to that of the parent T-2 whereas neosolaniol and deepoxyT-2 would be respectively 10 and 400 times less toxic than T-2 (Swanson et al., 1988).
Little information is available regarding biotransformation pathways of diacetoxyscirpenol (DAS). However, similar to T-2, this mycotoxin has been described to be rapidly and extensively metabolized in mammals and particularly in pigs (Bauer, 1995; Bauer et al., 1985). In this animal species, measurable amounts of DAS, monoacetoxyscirpenol (MAS) and scirpentriol were present in the blood for only 24 h after oral administration of DAS. These results indicate that the parent toxin would mainly undergo deacetylation at the C-4 and C-15 positions in the animal body. As a similar 15-deacetylation of DAS was observed using ovine rumen bacteria (Matsushima et al., 1996), so intestinal microflora in rats, cattle and swine completely biotransformed DAS to deacetylated deepoxidation products like deepoxy MAS and deepoxy scirpentriol. In contrast, faecal microflora from chickens, horses and dogs failed to reduce the epoxide groups in DAS and yielded only the deacylation products (Swanson et al., 1988). In terms of toxicological properties, MAS or scirpentriol are considered deleterious metabolites whereas both glucuronidation and epoxide reduction would be significant detoxification reaction steps for trichothecene mycotoxins.
Even though deoxynivalenol (DON) appears less toxic when compared to T-2 or DAS, its metabolism has been investigated in various animal species. After a preliminary study demonstrating the glucuronidation of DON and its long elimination halflife in sheep, Prelusky et al. (1986) defined the excretion profile of the same toxin administered to sheep. Following oral administration, urinary excretion products were DON, conjugated DON and conjugated deepoxyDON. These urinary metabolites accounted respectively for 2.1, 3.6 and 1.2% of the dose whereas between 54 and 75% of the oral dose were recovered in feces as unchanged DON (34-46%) or deepoxyDON (20-29%). In lactating cows, glucuronidation of deepoxyDON appeared to be one of the major metabolic pathways of this mycotoxin (Cote et al., 1986). Actually, deepoxidation of DON has been demonstrated to be an important hydrolytic metabolism influenced also by bovine rumen microorganisms or intestinal microflora of chickens (He et al., 1992).
ZEARALENONE
Zearalenone (ZEN), an oestrogenic metabolite produced by Fusarium spp., has a versatile effect on fertility of various breeding animals. After preliminary results demonstrating that free ZEN was the predominant compound found in the urine of rats, Mirocha et al. (1981) investigated the metabolic pattern of ZEN in the urine of various species including man. Both free and conjugated ZEN, α- and ß-zearalenols accounted for 63, 32 and 5% in urine of pigs. In cow urine, all three compounds were recovered but glucuronide and sulfate conjugates of ß-zearalenol were the predominant metabolites. In man, ZEN and α-zearalenol were the major metabolites followed by ß-zearalenol; all of which were in the glucuronide form. Both these reductive and conjugative metabolic pathways were confirmed by using in vitro models such as rat liver homogenate (Kiessling and Pettersson, 1978) or sow intestinal mucosa (Olsen et al., 1987). After characterization of the enzyme responsible for liver ZEN reductase activity, Ueno et al. (1983) described interspecies differences in these enzymes and indicated the presence of two distinct types of ZEN reductases differing in optimal pH whereas the stereospecific reduction of ZEN would depend on animal species.
Interestingly, similar reductive pathways of ZEN to α-zearalenol and to a lesser degree to ßzearalenol were described using intact rumen fluid or fractions of rumen protozoa and bacteria from sheep or cattle (Kiessling et al., 1984). In terms of toxicity, all these results concerning ZEN metabolism would be of major toxicological significance since the oestrogenic activity of α-zearalenol would be about 10 times greater than that of ZEN. Moreover, interspecies differences in α-reduction of ZEN could explain the large differences in oestrogenic properties developed by this mycotoxin among the various animal species, particularly between rabbits and poultry (Pompa et al., 1986).
FUMONISIN B1
When investigated in rats, the fate of radiolabelled fumonisin B1 (FB1) administered orally (Shephard et al., 1992) clearly showed the limited absorption of this mycotoxin. Consequently, only trace amounts were recovered in urine, suggesting that a large part does not reach general circulation due to either low absorption or rapid biliary elimination during the first pass through the liver following gut absorption. A complementary investigation using primary rat hepatocyte cultures (Cawood et al., 1994) indicated that no metabolites were detected in the fractionation of the culture medium. Nor were they detected using microsomal enzyme preparations efficient in term of esterase or P450 monooxygenase activities even though this toxin has been described to inhibit sphingosine N-acyl-transferase in cultured hepatocytes. Recently, Shephard et al. (1994) investigated the fate of FB1 in non-human primates in which faecal excretion of radioactivity accounted for 61% of the administered dose and urinary excretion only 1.2%. In faeces, both the parent compound, two partially-hydrolysed monoester metabolites and the aminopentol were identified, indicating that FB1 is the substrate of hydrolases active either during its passage through the liver into bile or from intestinal enzymes or microorganisms.
HPLC analysis of urine failed to detect any hydrolysis products, whereas FB1 was described as poorly metabolised by bovine ruminal microflora (Caloni et al., 2000). Regarding metabolite toxicity, aminopentol was found to be less effective in disrupting sphingolipid biosynthesis in rat hepatocytes in comparison to the unchanged mycotoxin (Van der Westhuizen et al., 1998).
Conclusion
During the last two decades much experimental data has accumulated to allow a better understanding of the biological fate of mycotoxins and the ways in which metabolic processes determine ultimate toxicity of these toxins. Due to their oestrogenic, carcinogenic, immunotoxic or general toxicological properties, these naturally occurring contaminants represent risks for both human and animal health. Moreover, the transfer of mycotoxins or their metabolites to animal-derived food products such as meat and milk demonstrates the importance of research which allows thorough mycotoxin risk assessment. Nevertheless, the biological fate of toxins such as deoxynivalenol, fumonisin B1, patulin, T-2 toxin or rubratoxin is not yet completely understood.
References
Author: PIERRE GALTIERAoyama, T., S. Yamano, P.S. Guzelian, H.V. Gelboin and F.J. Gonzalez. 1990. Five of 12 forms of vaccinia virus-expressed human hepatic cytochrome P450 metabolically activate aflatoxin B. Proc. Natl. Acad. Sci., USA, 87:4790.
Ball, R.W. and R.A. Coulombe. 1991. Comparative biotransformation of aflatoxin Bl in mammalian airway epithelium. Carcinogenesis 12:305.
Ball, R.W., D.W. Wilson and R.A. Coulombe. 1990. Comparative formation and removal of aflatoxin BJ-DNA adducts in cultured mammalian tracheal epithelium. Cancer Res. 50:4918.
Battista, J.R. and L.J. Mamett. 1985. Prostaglandin H synthase-dependent epoxidation of aflatoxin Bl. Carcinogenesis 6:1227.
Bauer, J. 1995. The metabolism of trichothecenes in swine. Dtsh. Tierarztl. Wochenschr. 102:50.
Bauer, J., W. Bollwahn, M. Gareis, B. Gedek and K. Heinritzi. 1985. Kinetic profiles of diacetoxyscirpenol and two of its metabolites in blood serum of pigs. Appl. Environ. MicrobioL 49:842.
Caloni F., M. Spotti, H. Auerbach, H. Op den Camp, J. Fink Gremmels and G. Pompa. 2000. In vitro metabolism of fumonisin B1 by ruminal microflora. Vet. Res. Com. 24:379.
Campbell, T.C. and J.R. Hayes. 1976. The role of aflatoxin metabolism in its toxic lesion. Toxicol. Appl. Pharmacol. 35:199.
Castegnaro, M. and D. MacGregor. 1998. Carcinogenic risk assessment of mycotoxins. Rev. Med. Vet. 149:671.
Castegnaro, M., H. Bartsch, J.C. Bereziat, P. Arvela, J. Michelon and L. Broussolle. 1989. Polymorphic ochratoxin A hydroxylation in rat strains phenotyped as poor and extensive metabolizers of debrisoquine. Xenobiotica 19:225.
Cawood, M.E., W.C.A. Gelderblom, J.F. Alberts and S.D. Snyman. 1994. Interaction of C-labelled fumonisin B, mycotoxins with primary rat hepatocyte cultures. Fd. Chem. Toxicol. 32:627.
Chu, F.S., I. Noh and C.C. Chang. 1972. Structural requirements for ochratoxin intoxication. Life Sci. 11:503.
Coles, B., D.J. Meyer, B. Ketterer and C.A. Stanton. 1985. Studies on the detoxication of microsomally activated aflatoxin Bl by glutathione and glutathione transferases in vitro. Carcinogenesis 6:693.
Cote, L.M., A.M. Dahlem, T. Yoshizawa, S.P. Swanson and W.B. Buck. 1986. Excretion of deoxynivalenol and its metabolite in milk, urine and feces of lactating dairy cows. J. Dairy Sci. 69:2416.
Coulombe, R.A., D.W. Shelton, R.O. Sinnhuber and J.E. Nixon. 1982. Comparative mutagenicity of aflatoxins using a salmonella/trout hepatic enzyme activation system. Carcinogenesis 3:1261.
Creppy, E.E., F.C. Stormer, R. Roschenthaler and G. Dirheimer. 1983. Effects of two metabolites of ochratoxin A, (4R)-4-hydroxyochratoxin A and ochratoxin A, on immune response in mice. Inf. Immun. 39:1015.
Crespi, C.L., B.W. Penman, D.T. Steimel, H.V. Gelboin and F.J. Gonzalez. 1991. The development of a human cell line stably expressing human CYP3A4: role in the metabolic activation of aflatoxin B1 and comparison to CYP1A2 and CYP2A3. Carcinogenesis 12:355.
De Groene, E.M., I.G.A.M. Hassing, M.J. Blom, W. Seiner, J. Fink-Gremmels and G.J. Horbach. 1996. Development of human cytochrome P450- expressing cell lines: application in mutagenicity testing of ochratoxin A. Cancer Res. 56:299.
Eaton, D.L. and J.D. Groopman. 1994. The Toxicology of Aflatoxins: Human Health, Veterinary and Agricultural Significance. Academic Press, San Diego. Essigmann, J.M., R.G. Croy, R.A. Bennett and G.N.Wogan. 1982. Metabolic activation of aflatoxin B: Patterns of DNA adduct formation, removal and excretion in relation to carcinogenesis. Drug. Metab. Rev. 13:581.
Forrester, L.M., G.E. Neal, F.G. Judah, M.J. Glancey and C.R. Wolf. 1990. Evidence for involvement of multiple forms of cytochrome P450 in aflatoxin Bl metabolism in human liver. Proc. Natl. Acad. Sci. USA, 87:8306.
Gallagher, E.P., L.C. Wienkers, P.L. Stapleto, K.L. Kunze and P.L. Eaton. 1994. Role of human microsomal and human complementary DNA-expressed cytochromes P450 1A2 and P450 3A4 in the bioactivation of aflatoxin Bl. Cancer Res. 54:101.
Galtier P. 1974. Fate of ochratoxin A in the animal organism. II. Tissue distribution and elimination in the rat. Ann. Rech. Vet. 5:319.
Galtier, P. and M. Alvinerie. 1976. In vitro transformation of ochratoxin A by animal microbial floras. Ann. Rech. Vet. 7:91.
Galtier, P., J.L. Charpenteau, M. Alvinene and C. Labouche. 1979. The pharmacokinetic profile of ochratoxin A in the rat after oral and intravenous administration. Drug Metab. Dispos. 7:429.
Gareis, M., A. Hashem, J. Bauer and B. Gedek. 1986. Identification of glucuronide metabolites of T-2 toxin and diacetoxyscirpenol in the bile of isolated perfused rat liver. Toxicol. Appl. Pharmacol. 84:168.
Gopalan, P., D.E. Jensen and P.D. Lotlikar. 1992. Glutathione conjugation of microsome-mediated and synthetic aflatoxin B 1-8.9-oxide by purified glutathione-transferases from rats. Cancer Lett. 64:225.
Harrison, J.C. and R.C. Gamer. 1991. Immunological and HPLC detection of aflatoxin adducts in human tissues after an acute poisoning incident in S.E. Asia. Carcinogenesis 12:741.
Hayes, A.W. 1973. Mycotoxin teratogenicity and mutagenicity. CRC Press, Boca Raton. He, P., L.G. Young and C. Forsberg. 1992. Microbial transformation of deoxynivalenol. Appl. Environ. MicrobioL 58:3857.
Hutchinson, R.D. and P.S. Steyn. 1971. The isolation and structure of 4-hydroxyochratoxin A and 7-carboxy-3,4-dihydro-8-hydroxy-3- methylisocoumarin from Penicillium viridicatum. Tetrahedron Lett. 43:4033.
Johnsen, H., E. Odden, B.A. Johnsen and F. Fonnum. 1988. Metabolism of T-2 toxin by blood cell carboxylesterases. Biochem. Pharmacol. 37:3193.
Kiessling, K.H. and H. Pettersson. 1978. Metabolism of zearalenone in rat liver. Acta Pharmacol. Toxicol. 43:285.
Kiessling. K.H., H. Pettersson, K. Sandholm and M. Olsen. 1984. Metabolism of aflatoxin, ochratoxin, zearalenone and three trichothecenes by intact rumen fluid, rumen protozoa and rumen bacteria. Appl. Environ. MicrobioL 47:1070.
Le Bars, J. and P. Le Bars. 1996. Recent acute and subacute mycotoxicoses recognized in France. Vet. Res. 27:383.
Liu, L. and T.E. Massey. 1992. Bioactivation of aflatoxin Bl by lipoxygenases, prostaglandin H synthase and cytochrome P450 monooxygenase in Guinea pig tissues. Carcinogenesis 13:533.
Liu, L., J.M. Daniels, R.K. Stewart and T.E. Massey. 1990. In vitro prostaglandin H synthaseand monooxygenase-mediated binding of aflatoxin Bl to DNA in Guinea pig tissue microsomes. Carcinogenesis 11:1915.
Liu, Y.H., J. Taylor, P. Linko, G.W. Lucier and C.L. Thompson. 1991. Glutathione S-transferase p. in human lymphocyte and liver: role in modulating formation of carcinogen-derived DNA Aaducts. Carcinogenesis 12:2269.
Lutz, W.K., W. Jaggi, J. Luthy, P. Sagelsdorff and C. Schlatter. 1980. In vivo covalent binding of aflatoxin B and aflatoxin Ml to liver DNA of rat, mouse and pig. Chem. Biol. Interact. 32:249.
Matsushima, T., E. Okamoto, E. Miyagawa, Y. Matsui, H. Shimizu and E. Asano. 1996. Deacetylation of diacetoxyscirpenol to 15- acetoxyscirpenol by rumen bacteria. J. Gen. Appl. MicrobioL 42:225.
Mirocha. C.J., S.V. Pathre and T.S. Robison. 1981. Comparative metabolism of zearalenone and transmission into bovine milk. Fd. Cosmet. Toxicol. 19:25.
Neal, G.E. and J.A. Green. 1983. The requirement for glutathione S-transferase in the conjugation of activated aflatoxin Bl during aflatoxin hepatocarcinogenesis in the rat. Chem. Biol. Interact. 45:259.
Nel, W. and I.F.H. Purchase. 1968. The fate of ochratoxin A in rats. J.S. Afr. Chem. Inst. 21:87.
Olsen, M., H. Pettersson, K. Sandholm, A. Visconti and K.H. Kiessling. 1987. Metabolism of zearalenone by sow intestinal mucosa in vitro. Fd. Chem. Toxicol. 25:681.
Oswald, I.P. and C. Comera. 1998. Immunotoxicity of mycotoxins. Rev. Med. Vet. 149:585.
Pitout M.J. 1969. The hydrolysis of ochratoxin A by some proteolytic enzymes. Biochem. Pharmacol. 18:485.
Pompa, G., C. Montesissa, F.M. Di Lauro and L. Fadini. 1986. The metabolism of zearalenone in subcellular fractions from rabbit and hen hepatocytes and its estrogenic activity in rabbits. Toxicology 42:69.
Prelusky D.B., D.M. Veira, H.L. Trenholm and K.E. Hartin. 1986. Excretion profiles of the mycotoxin deoxynivalenol, following oral and intravenous administration to sheep. Fund. Appl. Toxicol. 6:356.
Raj, H.G. and P.D. Lotlikar. 1984. Urinary excretion of thiol conjugates of aflatoxin B1 in rats and hamsters. Cancer Lett. 22:125.
Shephard, G.S., P.G. Thiel, E.W. Sydenham, J.F. Alberts and W.C.A. Gelderblom. 1992. Fate of a single dose of the 14C-labelled mycotoxin, fumonisin B1, in rats. Toxicon. 30:768.
Shephard, G.S., P.G. Thiel, E.W. Sydenham, J.F. Alberts and M.E. Cawood. 1994. Distribution and excretion of a single dose of the mycotoxin fumonisin B1, in a non-human primate. Toxicon 32:735.
Shimada, T. and P.P. Guengerich. 1989. Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver. Proc. Natl. Acad. Sci. USA 86:462.
Stormer, F.C. and J.I. Pedersen. 1980. Formation of 4-hydroxyochratoxin A from ochratoxin A by rat liver microsomes. Appl. Environ. Microbiol. 39:971.
Stormer, F.C., C.E. Hansen, J.I. Pedersen, G. Hvistendahl and A.J. Aasen. 1981. Formation of (4R)- and (4S)-4-hydroxyochratoxin A from ochratoxin A by liver microsomes from various species. Appl. Environ. Microbiol. 42:1051.
Stormer, F.C., O. Storen, C.E. Hansen, J.I. Pedersen and A.J. Aasen. 1983. Formation of (4R)- and (4S)-4-hydroxyochratoxin A and 10- hydroxyochratoxin A from ochratoxin A by rabbit liver microsomes. Appl. Environ. Microbiol. 45:1183.
Swanson, S.P., C. Helaszek, W.B Buck, H.R. Rood and W.M. Haschek. 1988. The role of intestinal microflora in the metabolism of trichothecene mycotoxins. Fd. Chem. Toxicol. 26:823.
Ueno, Y., F. Tashiro and T. Kobayashi. 1983. Species differences in zearalenone reductase activity. Fd. Chem. Toxicol. 21:167.
Valsta, L.M., J.D. Hendricks and G.S. Bailey. 1988. The significance of glutathione conjugation for aflatoxin B1 metabolism in rainbow trout and Coho salmon. Fd. Chem. Toxicol. 26:129.
Van der Westhuizen, L., G.S. Shepard, S.D. Snyman, S. Abel, S. Swanevelder and W.C. Gelderblom. 1998. Inhibition of sphingolipid biosynthesis in rat primary hepatocyte cultures by fumonisin B1 and other structurally related compounds. Food Chem. Toxicol. 36:497.
Van Walbeek, W., C.A. Moodie, P.M. Scott, J. Harwig and H.C. Grice. 1971. Toxicity and excretion of ochratoxin A in rats intubated with pure ochratoxin A or fed cultures of Penicillium viridicatwn. Toxicol. Appl. Pharmacol. 20:439.
Wei, C.I., M.R. Marshall and D.P. Hsieh. 1985. Characterization of water-soluble glucuronide and sulphate conjugates of aflatoxin B1. 1. Urinary excretion in monkey, rat and mouse. Fd. Chem. Toxicol. 23:809.
Yagen, B. and M. Bialer. 1993. Metabolism of pharmacokinetics of T-2 toxin and related trichothecenes. Drug Metab. Rev. 25:281.
Yoschizawa, T., C.J. Mirocha, J.C. Behrens and S.P. Swanson. 1981. Metabolic fate of T-2 toxin in a lactating cow. Fd. Cosmet. Toxicol. 19:31.
Laboratoire de Pharmacologie-Toxicologie INRA, Toulouse, France





