Structure and immune recognition of the porcine epidemic diarrhea virus spike protein
Author details:
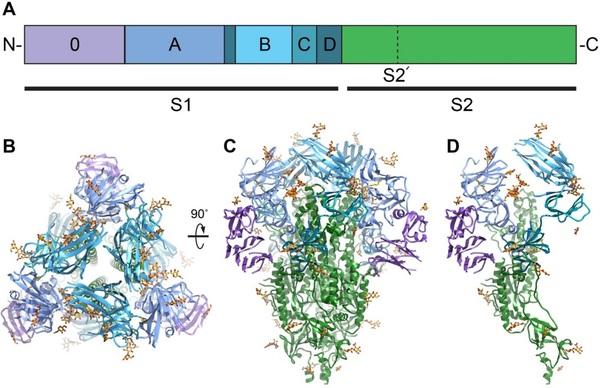
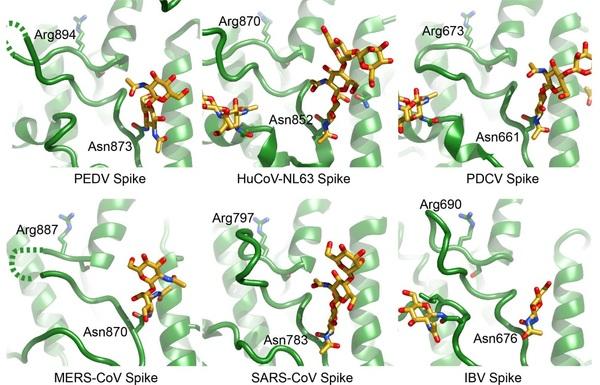
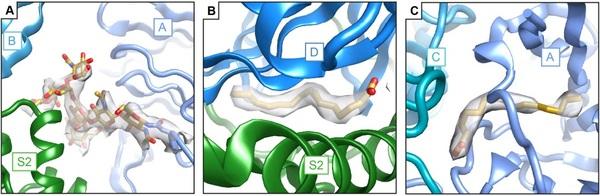
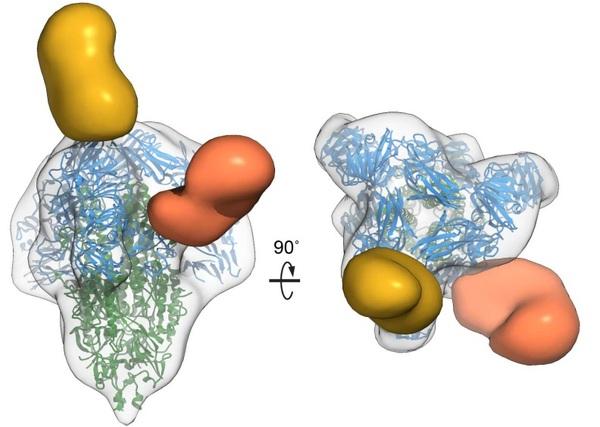
Porcine epidemic diarrhea virus is an alphacoronavirus responsible for significant morbidity and mortality in pigs. A key determinant of viral tropism and entry, the PEDV spike protein is a key target for the host antibody response and a good candidate for a protein-based vaccine immunogen. We used electron microscopy to evaluate the PEDV spike structure, as well as pig polyclonal antibody responses to viral infection. The structure of the PEDV spike reveals a configuration similar to that of HuCoV-NL63. Several PEDV protein-protein interfaces are mediated by non-protein components including a glycan at Asn264 and two bound palmitoleic acid molecules. The polyclonal antibody response to PEDV infection shows a dominance of epitopes in the S1 region. This structural and immune characterization provides new insights into coronavirus spike stability determinants and explores the immune landscape of viral spike proteins.
Adams, P.D., Afonine, P.V., Bunkoczi, G., Chen, V.B.,
Davis, I.W., Echols, N., Headd, J.J., Hung, L.W., Kapral,
G.J., Grosse-Kunstleve, R.W., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66,
213-221.
Barad, B.A., Echols, N., Wang, R.Y., Cheng, Y., DiMaio,
F., Adams, P.D., and Fraser, J.S. (2015). EMRinger: side chain-directed model and map validation for 3D cryoelectron microscopy. Nat Methods 12, 943-946.
Belouzard, S., Chu, V.C., and Whittaker, G.R. (2009).
Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc
Natl Acad Sci U S A 106, 5871-5876.
Berndsen, Z., Bowman, C., Jang, H., and Ward, A.B. (2017). EMHP: an accurate automated hole masking algorithm for single-particle cryo-EM image processing.
Bioinformatics 33, 3824-3826.
Bianchi, M., Turner, H.L., Nogal, B., Cottrell, C.A., Oyen,
D., Pauthner, M., Bastidas, R., Nedellec, R., McCoy,
L.E., Wilson, I.A., et al. (2018). Electron-MicroscopyBased Epitope Mapping Defines Specificities of
Polyclonal Antibodies Elicited during HIV-1 BG505
Envelope Trimer Immunization. Immunity 49, 288-
300.e288.
Bosch, B.J., van der Zee, R., de Haan, C.A., and Rottier, P.J. (2003). The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77, 8801-8811.
Bullough, P.A., Hughson, F.M., Skehel, J.J., and Wiley,
D.C. (1994). Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37-43.
Chambers, P., Pringle, C.R., and Easton, A.J. (1990).
Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J Gen Virol 71 ( Pt 12), 3075-3080.
Chang, S.H., Bae, J.L., Kang, T.J., Kim, J., Chung, G.H.,
Lim, C.W., Laude, H., Yang, M.S., and Jang, Y.S. (2002).
Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells 14, 295-299.
Diep, N.V., Norimine, J., Sueyoshi, M., Lan, N.T., and
Yamaguchi, R. (2017). Novel Porcine Epidemic Diarrhea
Virus (PEDV) Variants with Large Deletions in the Spike (S) Gene Coexist with PEDV Strains Possessing an Intact
S Gene in Domestic Pigs in Japan: A New Disease
Situation. PLoS One 12, e0170126.
Emsley, P., Lohkamp, B., Scott, W.G., and Cowtan, K. (2010). Features and development of Coot. Acta
Crystallogr D Biol Crystallogr 66, 486-501.
Hoang, H., Killian, M.L., Madson, D.M., Arruda, P.H., Sun,
D., Schwartz, K.J., and Yoon, K.J. (2013). Full-Length
Genome Sequence of a Plaque-Cloned Virulent Porcine
Epidemic Diarrhea Virus Isolate (USA/Iowa/18984/2013) from a Midwestern U.S. Swine
Herd. Genome Announc 1.
Hou, Y., Lin, C.M., Yokoyama, M., Yount, B.L., Marthaler,
D., Douglas, A.L., Ghimire, S., Qin, Y., Baric, R.S., Saif,
L.J., et al. (2017). Deletion of a 197-Amino-Acid Region in the N-Terminal Domain of Spike Protein Attenuates
Porcine Epidemic Diarrhea Virus in Piglets. J Virol 91.
Julien, J.P., Cupo, A., Sok, D., Stanfield, R.L., Lyumkis, D.,
Deller, M.C., Klasse, P.J., Burton, D.R., Sanders, R.W.,
Moore, J.P., et al. (2013). Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342, 1477-1483.
Kirchdoerfer, R.N., Cottrell, C.A., Wang, N., Pallesen, J.,
Yassine, H.M., Turner, H.L., Corbett, K.S., Graham, B.S.,
McLellan, J.S., and Ward, A.B. (2016). Pre-fusion structure of a human coronavirus spike protein. Nature
531, 118-121.
Kirchdoerfer, R.N., Wang, N., Pallesen, J., Wrapp, D.,
Turner, H.L., Cottrell, C.A., Corbett, K.S., Graham, B.S.,
McLellan, J.S., and Ward, A.B. (2018). Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis.
Sci Rep 8, 15701.
Lee, C. (2015). Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol J
12, 193.
Li, B.X., Ge, J.W., and Li, Y.J. (2007). Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology 365, 166-172.
Li, C., Li, W., Lucio de Esesarte, E., Guo, H., van den Elzen,
P., Aarts, E., van den Born, E., Rottier, P.J.M., and Bosch,
B.J. (2017a). Cell Attachment Domains of the Porcine
Epidemic Diarrhea Virus Spike Protein Are Key Targets of Neutralizing Antibodies. J Virol 91.
Li, W., Luo, R., He, Q., van Kuppeveld, F.J.M., Rottier,
P.J.M., and Bosch, B.J. (2017b). Aminopeptidase N is not required for porcine epidemic diarrhea virus cell entry.
Virus Res 235, 6-13.
Li, W., van Kuppeveld, F.J.M., He, Q., Rottier, P.J.M., and
Bosch, B.J. (2016). Cellular entry of the porcine epidemic diarrhea virus. Virus Res 226, 117-127.
Li, Z., Tomlinson, A.C., Wong, A.H., Zhou, D., Desforges,
M., Talbot, P.J., Benlekbir, S., Rubinstein, J.L., and Rini,
J.M. (2019). The human coronavirus HCoV-229E Sprotein structure and receptor binding. Elife 8.
Lyumkis, D., Julien, J.P., de Val, N., Cupo, A., Potter, C.S.,
Klasse, P.J., Burton, D.R., Sanders, R.W., Moore, J.P.,
Carragher, B., et al. (2013). Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer.
Science 342, 1484-1490.
Madson, D.M., Magstadt, D.R., Arruda, P.H., Hoang, H.,
Sun, D., Bower, L.P., Bhandari, M., Burrough, E.R.,
Gauger, P.C., Pillatzki, A.E., et al. (2014). Pathogenesis of porcine epidemic diarrhea virus isolate (US/Iowa/18984/2013) in 3-week-old weaned pigs. Vet
Microbiol 174, 60-68.
Masuda, T., Murakami, S., Takahashi, O., Miyazaki, A.,
Ohashi, S., Yamasato, H., and Suzuki, T. (2015). New porcine epidemic diarrhoea virus variant with a large deletion in the spike gene identified in domestic pigs.
Arch Virol 160, 2565-2568.
Millet, J.K., and Whittaker, G.R. (2015). Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res 202, 120-134.
Okda, F.A., Lawson, S., Singrey, A., Nelson, J., Hain, K.S.,
Joshi, L.R., Christopher-Hennings, J., Nelson, E.A., and
Diel, D.G. (2017). The S2 glycoprotein subunit of porcine epidemic diarrhea virus contains immunodominant neutralizing epitopes. Virology 509, 185-194.
Pallesen, J., Wang, N., Corbett, K.S., Wrapp, D.,
Kirchdoerfer, R.N., Turner, H.L., Cottrell, C.A., Becker,
M.M., Wang, L., Shi, W., et al. (2017). Immunogenicity and structures of a rationally designed prefusion MERSCoV spike antigen. Proc Natl Acad Sci U S A 114, E7348- e7357.
Park, J.E., Li, K., Barlan, A., Fehr, A.R., Perlman, S.,
McCray, P.B., Jr., and Gallagher, T. (2016). Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad
Sci U S A 113, 12262-12267.
Pensaert, M.B., and de Bouck, P. (1978). A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58, 243-247.
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S.,
Greenblatt, D.M., Meng, E.C., and Ferrin, T.E. (2004).
UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605-1612.
Pickett, B.E., Sadat, E.L., Zhang, Y., Noronha, J.M.,
Squires, R.B., Hunt, V., Liu, M., Kumar, S., Zaremba, S.,
Gu, Z., et al. (2012). ViPR: an open bioinformatics database and analysis resource for virology research.
Nucleic Acids Res 40, D593-598.
Punjani, A., Rubinstein, J.L., Fleet, D.J., and Brubaker,
M.A. (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat
Methods 14, 290-296.
Sawyer, W., and Sherwell, P. (2014). This Little Piggy
Cried P-E-D-v All the Way Home: Estimating the Imacts of PEDv in North America. Rabobank AgFocus, 8.
Shang, J., Zheng, Y., Yang, Y., Liu, C., Geng, Q., Luo, C.,
Zhang, W., and Li, F. (2018a). Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog 14, e1007009.
Shang, J., Zheng, Y., Yang, Y., Liu, C., Geng, Q., Tai, W.,
Du, L., Zhou, Y., Zhang, W., and Li, F. (2018b). CryoElectron Microscopy Structure of Porcine
Deltacoronavirus Spike Protein in the Prefusion State. J
Virol 92.
Song, W., Gui, M., Wang, X., and Xiang, Y. (2018). CryoEM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog
14, e1007236.
Stevenson, G.W., Hoang, H., Schwartz, K.J., Burrough,
E.R., Sun, D., Madson, D., Cooper, V.L., Pillatzki, A.,
Gauger, P., Schmitt, B.J., et al. (2013). Emergence of
Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet
Diagn Invest 25, 649-654.
Suloway, C., Pulokas, J., Fellmann, D., Cheng, A., Guerra,
F., Quispe, J., Stagg, S., Potter, C.S., and Carragher, B. (2005). Automated molecular microscopy: the new
Leginon system. J Struct Biol 151, 41-60.
Sun, D.B., Feng, L., Shi, H.Y., Chen, J.F., Liu, S.W., Chen,
H.Y., and Wang, Y.F. (2007). Spike protein region (aa
636789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol 51, 149-
156.
Taguchi, F., and Matsuyama, S. (2002). Soluble receptor potentiates receptor-independent infection by murine coronavirus. J Virol 76, 950-958.
Thirstrup, K., Carriere, F., Hjorth, S., Rasmussen, P.B.,
Woldike, H., Nielsen, P.F., and Thim, L. (1993). One-step purification and characterization of human pancreatic lipase expressed in insect cells. FEBS Lett 327, 79-84.
Tortorici, M.A., Walls, A.C., Lang, Y., Wang, C., Li, Z.,
Koerhuis, D., Boons, G.J., Bosch, B.J., Rey, F.A., de
Groot, R.J., et al. (2019). Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct
Mol Biol 26, 481-489.
Walls, A.C., Tortorici, M.A., Bosch, B.J., Frenz, B., Rottier,
P.J.M., DiMaio, F., Rey, F.A., and Veesler, D. (2016a).
Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114-117.
Walls, A.C., Tortorici, M.A., Frenz, B., Snijder, J., Li, W.,
Rey, F.A., DiMaio, F., Bosch, B.J., and Veesler, D. (2016b). Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol 23, 899-905.
Walls, A.C., Tortorici, M.A., Snijder, J., Xiong, X., Bosch,
B.J., Rey, F.A., and Veesler, D. (2017). Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad
Sci U S A 114, 11157-11162.
Walls, A.C., Xiong, X., Park, Y.J., Tortorici, M.A., Snijder,
J., Quispe, J., Cameroni, E., Gopal, R., Dai, M.,
Lanzavecchia, A., et al. (2019). Unexpected Receptor
Functional Mimicry Elucidates Activation of Coronavirus
Fusion. Cell 176, 1026-1039.e1015.
Wang, R.Y., Song, Y., Barad, B.A., Cheng, Y., Fraser, J.S., and DiMaio, F. (2016). Automated structure refinement of macromolecular assemblies from cryo-EM maps using
Rosetta. Elife 5.
Williams, C.J., Headd, J.J., Moriarty, N.W., Prisant, M.G.,
Videau, L.L., Deis, L.N., Verma, V., Keedy, D.A.,
Hintze, B.J., Chen, V.B., et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293-315.
Wilson, I.A., Skehel, J.J., and Wiley, D.C. (1981). Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289, 366-373.
Wood, E.N. (1977). An apparently new syndrome of porcine epidemic diarrhoea. Vet Rec 100, 243-244.
Wrapp, D., and McLellan, J.S. (2019). The 3.1 A cryo-EM structure of the porcine epidemic diarrhea virus spike protein in the prefusion conformation. J Virol.
Xiong, X., Tortorici, M.A., Snijder, J., Yoshioka, C., Walls,
A.C., Li, W., McGuire, A.T., Rey, F.A., Bosch, B.J., and
Veesler, D. (2018). Glycan Shield and Fusion Activation of a Deltacoronavirus Spike Glycoprotein Fine-Tuned for
Enteric Infections. J Virol 92.
Yuan, Y., Cao, D., Zhang, Y., Ma, J., Qi, J., Wang, Q., Lu,
G., Wu, Y., Yan, J., Shi, Y., et al. (2017). Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8, 15092.
Zhang, K. (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1-12.
Zheng, S.Q., Palovcak, E., Armache, J.P., Verba, K.A.,
Cheng, Y., and Agard, D.A. (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14,
331-332.
Zivanov, J., Nakane, T., Forsberg, B.O., Kimanius, D.,
Hagen, W.J., Lindahl, E., and Scheres, S.H. (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7.







.jpg&w=3840&q=75)