Introduction
Porcine circoviruses (PCVs) are members of the genus Circovirus included in the family Circoviridae, which comprises small icosahedral non-enveloped viruses with a circular single-stranded DNA (ssDNA) genome. There are two types of PCV, namely PCV1 and PCV2. PCV1 is a non-cytopathic contaminant of the PK15 cell line that was first identified in 1974 and described as non-pathogenic for pigs (Tischer et al., 1974; Allan et al., 1995). PCV2 is recognized as the essential infectious agent of post- eaning multisystemic wasting syndrome (PMWS; Ellis et al., 1998). This multifactorial disease causes severe growth retardation and weight loss and can prove fatal to nursery and fattening pigs (Segalés et al., 2005). More recently, the terminology of porcine circovirus disease (PCVD) has been adopted as a collective term to describe several disease complexes associated with PCV2 infection in pigs (Allan et al., 2002).
Difficulties associated with the consistent reproduction of PMWS under experimental conditions (Tomás et al., 2008) have hampered attempts to conduct a chronological assessment of PCV2 replication in vivo, necessitating the use of appropriate in-vitro cell culture models.
It has been reported that the attachment of PCV2 virus-like particles (VLPs) to the plasma membrane of monocytes/macrophages is determined by saturation of the receptors within the first 15 min of incubation (Misinzo et al., 2005). It is also known that PCV2 can be internalized by monocytes/macrophages in an in-vitro acidic environment and that this is an actine cholesterol dependent process (Misinzo et al., 2005), similar to that of dendritic (Vincent et al., 2007) and PK15 (Misinzo et al., 2009) cells. However, the small GTPase-dependent regulated clathrin- and caveolin-independent pathway appears to be more effective for PCV2 internalization, after which PCV2 would be transported from endosomes into lysosomes (Misinzo et al., 2009).The first stages of PCV2 replication in the cytoplasm have not been identified; however, it is known that the viral-positive ssDNA is converted into a double-stranded DNA replicative form (dsDNA) in the nucleus. Replicase (Rep and Rep0) and capsid (Cap) proteins form protein complexes in the cytoplasm and enter the nucleus (Mankertz et al., 2004; Meerts et al., 2005).
Following PCV2 DNA synthesis, the viral components appear to assemble in the nucleus and subsequent progeny virions can be detected in the cytoplasm (Meerts et al., 2005). These authors determined that the PCV2 replication cycle in PK15 cells takes 24e36 h, although there is a lack of information regarding the final stages of the process. Immunofluorescence studies conducted with PCV2-infected PK15 cells reported the detection of capsid protein in the perinuclear region of cytoplasm at 6e12 h post-inoculation (hpi; Cheung and Bolin, 2002; Meerts et al., 2005). These authors also reported viral capsid protein in the nucleus at 12-24 hpi, in perinuclear areas at 24e48 hpi and finally in the cytoplasm after 35 hpi.
Ultrastructural findings related to PCV2 infection have been described in lymph node histiocytes of PMWS- ffected pigs. In such studies, large numbers of intracytoplasmic inclusion bodies (ICIs) consisting of VLPs arranged in paracrystalline arrays were observed.
The existence of viral factories (VFs) was also reported. In some cases VFs were associated with mitochondria, suggesting that this organelle may play an important role in PCV2 replication (Rodríguez-Cariño and Segalés, 2009; Rodríguez- Cariño et al., 2010). However, to the best of authors´ knowledge, a complete ultrastructural study of PCV2 morphogenesis in cell culture has never been performed and only a single ultrastructural description has been published for PCV1 (Stevenson et al., 1999). Therefore, the purpose of the present work was to study the morphogenesis of PCV2 infection in a clone derived from the L35 lymphoblastoid cell line. Electron microscopy (EM) techniques were used to identify the cellular localization of viral particles and detect subcellular alterations at different time points post-infection.
Materials and Methods
Experimental Design
A clone derived from the L35 lymphoblastoid cell line (Kaeffer et al., 1990) was selected as a candidate host cell model for the morphological study of PCV2 infection as it has demonstrated a significantly higher level of PCV2 infection in comparison with other cell lines. In the present study, cultures of the L35-derived clone (supplied by F. McNeilly and G. Allan, AFBI, Stormont, Northern Ireland) were infected with PCV2 at a multiplicity of infection (MOI) of 10; mock-infected cell cultures were treated in a similar fashion with a PCV2-free L35 cell lysate. PCV2- and mock-infected cell cultures were incubated for 2 h at 37�C in 5% (v/v) CO2 atmosphere prior to centrifugation at 1,022 g for 10 min. The cells were washed by resuspending in 45 ml of fresh L35 medium (RPMI 1640 medium; Invitrogen, Paisley, UK), 2 mM glutamine, gentamycin (100 μg/ml), 1 mMsodium pyruvate and 15% (v/v) New Zealand fetal calf serum (PAA Laboratories, Pasching, Austria), followed by centrifugation as described previously. Cells were subjected to five separate washes. Mock and infected cell cultures were resuspended in L35 medium and divided between seven cell culture flasks. At each time point (0, 6, 12, 24, 48, 60 and 72 hpi), a flask of virus-infected cell culture and a flask of control cell culture were each divided between two centrifuge tubes and cell pellets were collected by centrifugation at 1,022 g for 10 min. One set of samples was processed for conventional transmission electron microscopy (TEM) to detect ultrastructural alterations due to virus infection, while the duplicate set of samples was prepared for localization of virus by means of immunogold labelling for TEM. The culture was checked by immunofluorescence at 48 hpi and the rate of infected cells was estimated to be 20-30%.
Ultrastructural Evaluation
Pellets of PCV2 infected and control samples were resuspended in a fixative solution of 2%(w/v) paraformaldehyde and 2.5% (v/v) glutaraldehyde (EM grade, Merck, Darmstadt, Germany) in phosphate buffer (PB; 0.1M, pH 7.4; Sigma-Aldrich, Steinheim, Germany) and placed on a rocking platform for 2 h. Following fixation, the cells were washed with PB followed by centrifugation at 1,278 g for 10 min. The supernatant was discarded and the pellets resuspended in 1% (w/v) paraformaldehyde prepared in PB. The samples were centrifuged at 7,676 g for 5 min and rinsed three times with PB. Then, cell culture pellets were post-fixed for 2 h with 1% (w/v) osmium tetroxide (TAAB Lab., UK) containing 0.8% (w/v) potassium hexocyanoferrate (Sigma-Aldrich, Steinheim, Germany) prepared in PB, followed by four washes with deionized water and sequential dehydration in acetone. All procedures were performed at 4�º�C.
Samples were embedded in Eponate 12�TM resin (Ted Pella Inc., Redding, California) and polymerized at 60º�C for 48 h. Semithin sections (1 μm thick) were obtained with a Leica ultracut UCT microtome (Leica Microsystems GmbH, Wetzlar, Germany), stained with 1% (w/v) aqueous toluidine blue solution and examined with a light microscope to identify areas with a sufficient cell concentration for further analysis. Ultrathin sections (70 nm in thickness) were cut with a diamond knife (45�, Diatome, Biel, Switzerland), placed on non-coated 200 mesh copper grids and contrasted with conventional uranyl acetate (30 min) and Reynolds lead citrate (5 min) solutions. Sections were observed with a Jeol 1400 transmission electron microscope (Jeol Ltd, Tokyo, Japan) equipped with a Gatan Ultrascan ES1000 CCD Camera.
The dead cell percentage was quantified by counting the number of total cells in at least three sections at each post-fixation time (approximately 100 cells/hpi).
Immunogold Labelling
Cell pellets were resuspended in fixative solutions of 4% (w/v) paraformaldehyde and 0.1% (v/v) glutaraldehyde (EM grade, Merck) in 0.1 M PB (Sigma-Aldrich) and placed on a rocking platform for 2 h. Following fixation, the cells were washed with PB followed by centrifugation at 1,278 g for 10 min. The cells were resuspended in1%(w/v) paraformaldehyde prepared in PB and centrifuged at 7,676 g for 5 min. Cell culture pellets were washed four times in PB, subsequently with 20 mM glycine and cryoprotected in a graded series of sucrose (0.7, 1.4 and 2.3 M; Sigma-Aldrich) solutions prepared in PB. All steps were performed at 4�C. Samples were snap frozen in liquid propane, immersed in methanol (Merck) containing 0.5 uranyl acetate for 72 h at �90�C, washed with methanol and embedded in Lowicryl HM20 resin (Polysciences Inc., Warrington, USA). Resin polymerization was performed with UV rays for 48 h at �45�C followed by 48 h at 25�C. The dehydration and substitution processes were performed in a Leica EMAFS automatic freeze substitution system (Wetzlar, Germany). Semithin (1 μm) and ultrathin (90 nm) sections were prepared with a Leica Ultracut UCT microtome (Leica Microsystems GmbH). Ultrathin sections were placed on gold grids (200 mesh) without a film layer.
Immunolabelling of PCV2 was performed using a PCV2 capsid protein monoclonal antibody, anti- Cap IgG2a (Ingenasa, Madrid, Spain). The sections in gold grids were treated for antigen retrieval with 10 mM citrate buffer pH 6.0 for 10 min at 95�C, refreshed for 15 min at room temperature (RT) and then washed twice with deionized water and twice with phosphate buffer saline (PBS). Grids were incubated for 30 min in 1% (w/v) bovine serum albumin (BSA; Sigma-Aldrich) in PBS (BSA/PBS). The sections were incubated with the primary antibody at a 1 in 5 dilution in 1% (w/v) BSA/PBS at 4�C overnight in a humidified chamber. The grids were washed with 1% (w/v) BSA/PBS and incubated with a secondary anti-mouse antibody coupled to 10 nm gold particles (British BioCell International, Cardiff, UK) in 1% (w/v) BSA/PBS for 40 min at RT. Grids were washed four times with PBS followed by five washes with deionized water. Sections treated with BSA/PBS instead of primary antibody served as negative controls. All grids were contrasted with uranyl acetate and Reynolds lead citrate solutions for 15 min and 1 min, respectively. Sections were examined with a Jeol 1400 transmission electron microscope (Jeol Ltd, Tokyo, Japan) equipped with a Gatan Ultrascan ES1000 CCD Camera.
Results
Lymphoblastoid cells infected with PCV2 were studied at different hpi. Results are presented as observations made in PCV2-inoculated cells compared with non- CV2- noculated control cells. Immunogold labelling was not performed in cell cultures at 0, 6 and 12 hpi due to insufficient cell concentrations in the ultrathin sections at these time points (less than five cells visualized per time point).
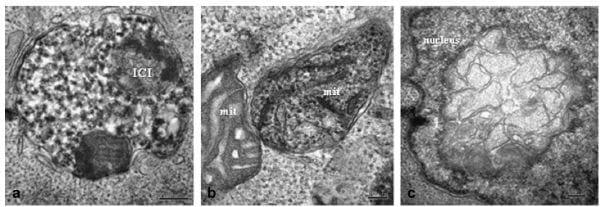
At 0 hpi, very few virus particles were observed, either in close proximity to the cell membrane or as intracellular aggregates in PCV2-infected cells. In some cells, VLPs were observed as organized double membrane- bound intracytoplasmic inclusion bodies (ICIs) located in the cytoplasm close to the plasma membrane and adjacent or proximal to mitochondria (Fig. 1a-c). These ICIs were 0.4-0.5 μm in diameter and contained recognizable VLPs approximately 17 nm in diameter. Some ICIs were observed as myelinoid figures forming large and complex electron dense bodies. At this time point, clear vesicular structures, which were absent in mock-infected cells, were also observed. These structures appeared to have a single membrane (although small stretches of double membrane were detected occasionally). The vesicles were round or oval in shape and often displayed an electron dense margin (Fig. 1d). These vesicles were associated with the rough endoplasmic reticulum (RER). At 0 hpi, 9-10% of all PCV2-infected cells showed recognizable VLPs. The number of dead cells in the PCV2-inoculated cell culture was 5e8% and was similar to the mock- nfected control. Cell viability in PCV2-infected cultures was 92-95% and was similar to the mock-infected control cells.
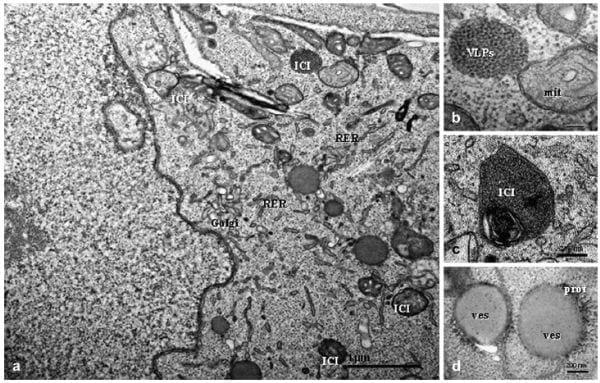
Fig. 1. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 0 hpi. (a) Recognizable intracytoplasmic inclusions (ICIs) surrounded by a double membrane and approximately 0.5 μm in diameter are distributed randomly throughout the cytoplasm; the Golgi complex and rough endoplasmic reticulum (RER) are slightly dilated. Bar, 1 μm. (b) ICI located next to mitochondrion (mit) that contains virus-like particles (VLPs) of approximately 17-18 nm diameter. Bar, 100 nm. (c) Some cells contain ICIs with double or triple membranes and internal myelinoid figures. Bar, 200 nm. (d) Cytoplasmic round vesicles (ves) are more frequently observed in infected cells; these vesicles have clear and homogeneous composition and are limited by an electron dense zone consistent with protein (prot). Bar, 200 nm.
At 6 hpi, PCV2-infected cells contained larger ICIs of 1 μm diameter, double membrane bound and with membrane debris inside (Fig. 2a). Some mitochondria containing VLPs of 18-20 nm in diameter were identified (Fig. 2b). Occasionally, the nuclear membrane had areas formed of large multimembrane structures (Fig. 2c). The percentage of cells containing VLPs increased at this time point post-inoculation, with VLPs detected in 12-14% of the cells examined. The number of dead cells was similar to that at 0 hpi in both PCV2-inoculated and mock-infected cells. Cell viability was found to be similar to that of viral and mock-infected cultures at 0 hpi.
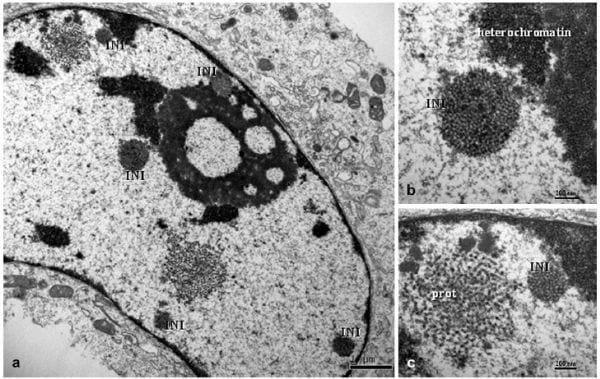
Non-membrane bound intranuclear inclusion bodies (INIs) were observed in infected cultures at 12 hpi. These INIs contained VLPs of 18-20 nm diameter (Fig. 3a) with an inter-particle distance of approximately 12-15 nm located adjacent to heterochromatin and the nuclear membrane (Fig. 3b). The INIs were circular in shape and approximately 0.4-0.6 μm in diameter. Loosely organized protein-like structures were observed as large nets of 1 μm diameter (Fig. 3c) in close proximity to the INIs within the nucleus. In the cytoplasm, ICIs similar to those described at previous time points were observed. The Golgi complex appeared slightly dilated in some infected cells. VLPs were observed in 18-20% of PCV2-inoculated cells. The number of dead cells in the PCV2-inoculated culture was 15-18%, whereas it was similar to 0 and 6 hpi for the mock-infected control.
At 24 hpi, some cells in the PCV2-infected cultures contained large INIs with VLPs (18-21 nm diameter), arranged in paracrystalline arrays, occupying large areas of the nucleus (Fig. 4a). In other cells, INIs contained VLPs with a granular appearance and were located close to nuclear membrane pores (Fig. 4b). These inclusion bodies were not detected by PCV2 capsid-specific immunogold labelling; however, some electron dense areas with irregular shapes that were proximal to INIs were labelled (Fig. 4c). Clusters of VLPs were observed next to the nuclear membrane (Fig. 4d), and showed immunolabelling of PCV2 Cap antigen (Fig. 4e). ICIs with VLPs were observed close to the nuclear membrane and attached to vesicles surrounded by electron dense areas. The inner and outer mitochondrial membranes showed clusters of colloidal gold particles (Fig. 4f). As observed at 12 hpi, the RER and Golgi complex appeared slightly dilated in almost all infected cells, although no evidence of PCV2 immunolabelling was detected in these organelles. INIs and/or ICIs with VLPs were found in 22-24% of PCV2-inoculated cells. The number of dead cells was 23-26% in the PCV2-inoculated culture; this value was similar to that of the previous sampling times for mock-infected cells (5-8%).
At 48 hpi, PCV2-infected cells contained several INIs similar in nature to those observed at 12 and24 hpi, but more numerous (Fig. 5a). Many INIs comprised VLPs (18-21 nm diameter) organized in oval or ring shapes. Some PCV2-infected cells showed several ICIs with VLPs (15 nm in diameter, with 10 nm between virus particles) arranged in paracrystalline arrays, surrounded by double membrane and containing whorled myelinoid figures (Fig. 5b). These ICIs were dispersed throughout the cytoplasm and usually contained high amounts of PCV2 antigen (Fig. 5c). However, most ICIs were adjacent to moderately dilated Golgi complexes and RER (Fig. 5d). Some ICIs comprising VLPs with electron dense cores were observed budding from the cytoplasmic cell membrane (Fig. 5e). The number of dead cells in the PCV2-infected cultures increased over time and constituted around 30% of the cell population studied at 48 hpi. This is in contrast to the percentage of dead cells in the mock-infected cells, which was approximately 8%. Some of the dead cells in the PCV2-infected cultures exhibited apoptotic nuclei (Fig. 5f) and a high number of VLPs (10-12 nm diameter) with an interparticle distance of 5 nm (Fig. 5f, inset). In some cases, VLPs were arranged in paracrystalline arrays with herringbone pattern, which were confirmed as PCV2 by immunogold labelling (Fig. 5g). Icosahedral virus particles were detected in 29-31% of PCV2- noculated cells.
A significant number of cells from the PCV2-infected cultures at 60 and 72 hpi showed INIs and ICIs with VLPs of 18-21 nm and 15-17 nm diameter, respectively (Fig. 6a). VLPs were observed in approximately 45% of PCV2-inoculated cells. PCV2 immunolabelling was similar to that observed at 24 and 48 hpi. INIs were not successfully labelled with the anti-PCV2 capsid monoclonal conjugate, but the nuclear membrane and non-specific adjacent areas were positively labelled. At these two latter time points, dilation of the Golgi complex was more evident in infected cells with several INIs and/or ICIs (Fig. 6b). ICIs showed recognizable viral structures (14-15 nm in diameter) arranged in paracrystalline arrays and surrounded by a membrane bilayer (Fig. 6c). At these time points post-inoculation around 40% of the cells in the PCV2-infected cultures were dead (Fig. 6d); some of them contained VLPs arranged in paracrystalline arrays, whichwere PCV2 positive by immunogold labelling (Fig. 6e). Dead cells accounted for 8-10%of the mock-infected cell culture.
The mock-infected cells were negative on PCV2- immunogold labelling. Additionally, no cells from the negative control sections (treated with BSA/PBS instead of primary antibody) were labelled.
Fig. 2. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 6 hpi. (a) Large and complex ICI with a double membrane, of granular composition with two rounded electron dense structures. Bar, 200 nm. (b) Twomitochondria (mit) are observed; the right one shows the matrix occupied by VLPs. Bar, 100 nm. (c) Nucleus containing a multimembrane structure. Bar, 0.2 μm.
Fig. 3. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 12 hpi. (a) Several intranuclear inclusion bodies (INIs) are observed; these are of 0.4-0.6 μm diameter and are not membrane bound. Bar, 1 μm. (b) Detail of the previous figure; the INI is located near to heterochromatin and contains VLPs 19 nm in diameter with 12-15 nm between particles. Bar, 200 nm. (c) An INI located next to heterochromatin and near to the nuclear membrane; an electron dense net consistent with protein aggregates (prot) is present to the left of the inclusion. Bar, 200 nm.
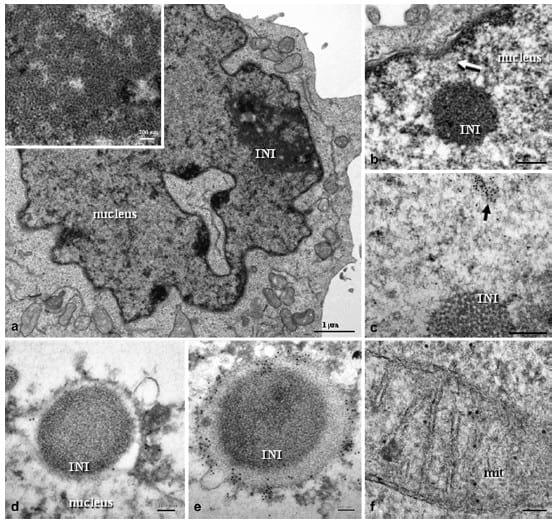
Fig. 4. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 24 hpi. (a) Large and irregular INI with VLPs arranged in paracrystalline arrays (insert at higher magnification). Bar, 1 μm. (b) INI with granular composition located in the proximity of a nuclear pore (white-arrow). Bar, 200 nm. (c) Colloidal gold particles were observed in an electron dense area (black-arrow) located near to a non-labelled INI composed of icosahedral VLPs. Bar, 200 nm. (d) INI budding from the nucleus, surrounded by nuclear membrane which shows colloidal gold particles. Bar, 100 nm. (e) Similar to the previous figure, in which the INI is almost totally bound by a heavily PCV2-gold-labelled nuclear membrane. Bar, 100 nm. (f) Mitochondrion showing PCV2-immunolabelled inner and outer membranes. Bar, 50 nm. (c)e(e) are immunogold labelling with PCV2 capsid protein monoclonal antibody and 10 nm gold particles.
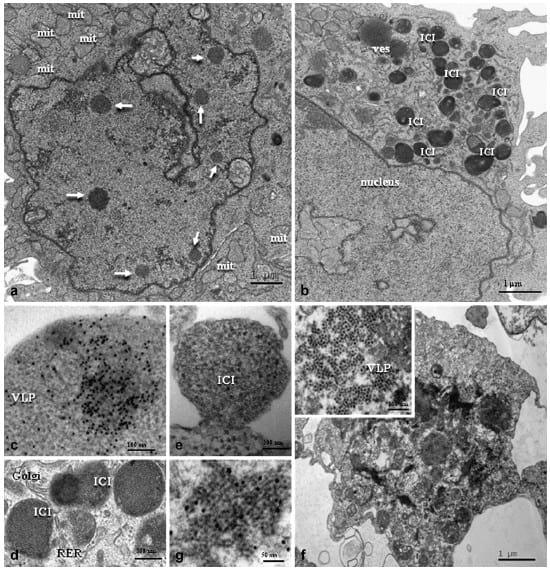
Fig. 5. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 48 hpi. (a) Nucleus containing a number of INIs; note the proliferation of swollen mitochondria (mit). Bar, 1 μm. (b) Numerous ICIs present in the cytoplasm. Bar, 1 μm. (c) Heavily PCV2-gold-labelled ICI showing icosahedral virions. Bar, 200 nm. (d) ICIs surrounded by a moderately dilated Golgi complex and rough endoplasmic reticulum (RER). Bar, 100 nm. (e) A cluster of VLPs with very electron dense core budding from the cell membrane. Bar, 200 nm. (f) Dead cell showing VLPs (insert at higher magnification) organized in paracrystalline arrays (VLPs of 10-12 nm diameter with 5 nmdistance between each). Bar, 100 nm. (g) VLPs organized as a herringbone pattern showing specific PCV2 immunolabelling. Bar, 50 nm. (c) and (g) are immunogold labelling with PCV2 capsid protein monoclonal antibody and 10 nm gold particles.
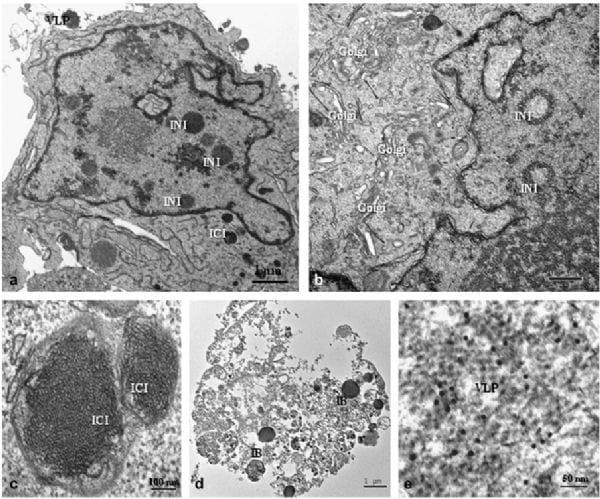
Fig. 6. PCV2-infected clone derived from the L35 lymphoblastoid cell line, 60-72 hpi. (a) Infected cell showing several INIs and ICIs containing VLPs as well as clusters of VLPs attached to the cytoplasmic membrane. Bar, 1 μm. (b) INIs in ring form within thenucleus; in addition, proliferation and dilation of the Golgi complex is seen. Bar, 0.5 μm. (c) Two ICIs surrounded by a double membrane with recognizable viral structures organized in paracrystalline arrays. Bar, 100 nm. (d) Dead cell with inclusion bodies (IB), including VLPs, arranged in paracrystalline arrays. Bar, 1 μm. (e) Free PCV2-immunogold-labelled icosahedral virions observed in the cytoplasm of the dead cell shown in (d). Bar, 50 nm. (e) is immunogold labelling with PCV2 capsid protein monoclonal antibody and 10 nm gold particles.
Discussion
This report describes the first ultrastructural study of PCV2 morphogenesis in cell culture. A clone derived from the L35 cell line was used to investigate the PCV2 replication cycle at the ultrastructural level because rates of infection in this cell line are higher than in other cell types (McNeilly et al., 2001). Since PCV2 is a DNA virus requiring DNA polymerase, cell lines with high division capabilities have been traditionally used for its culture; specifically, PK15 and swine kidney (SK) cell lines have been widely used for PCV research purposes (Stevenson et al., 1999; Cheung and Bolin, 2002; Meerts et al., 2005). In the study reported here, around 20-30% of lymphoblastoid cells were infected by PCV2 at 48 hpi, making the cell cloned from the L35 cell line highly suitable for conducting a complete morphogenesis study. Previous in-vivo and in-vitro studies have failed to demonstrate the association of PCV2 antigen/ nucleic acid with cells of a lymphocytic phenotype, but lymphocyte blast cells (L35) were found to be highly receptive to viral replication (McNair et al., 2005). These authors speculated that the numerous cycles of cell division of L35 cells provided an ideal environment for PCV2 replication.
The cell pellets collected at 0, 6 and 12 hpi were not able to be studied by means of immunogold methodology. Regrettably, even though ultrathin cuts suitable for the conventional electron microscopical study were obtained, the number of cells present in the ultrathin sections was too low (less than five per cut) for a reliable study. A number of reasons might explain this low cell availability in some samples. Most important is the lower number of cells present at these times post-inoculation compared with later times, since cell replication takes place over time. Furthermore, the use of paraformaldehyde fixative (more concentrated for immunogold studies) is associated with a loose cell pellet as compared with glutaraldehyde fixative (less concentrated for immunogold studies; C. Rodríguez- Cariño, personal observation). These two facts, together with the successive steps being performed at low temperature (cryotreatment, dehydration and embedding in resin), led to the partial loss of pellets in all cases and the almost total loss at the times post-inoculation with the least number of cells (0, 6 and 12 hpi).
On entry to the cell, the viral replication cycle involves an initial step within the cytoplasm, followed by the viral entrance into the nucleus and, finally, release of the virus to the cytoplasm before exiting the cell (Cheung and Bolin, 2002; Finsterbusch et al., 2005; Meerts et al., 2005; Misinzo et al., 2009). At the initial stage of PCV2 infection, the virus attaches to the cell surface and is internalized by endocytosis (Misinzo et al., 2009). In the present study, at time point 0 (i.e. 2 h after infection), clusters of VLPs were visualized randomly around the cells, close to the cell membrane. It was not surprising to find VLPs organized as ICIs even at this early time post-infection, as the virus had been in contact with the host cells for 2 h by this time. In the cytoplasm, VLPs were organized as double membrane bound inclusion bodies, which may result from engulfment of endocytic vesicles by lysosomes. These observations of the early stages of PCV2 internalization and cytoplasmic detection in the L35-derived clone are in agreement with those previously reported in PK15, SK, monocytes/macrophages, swine testicle (ST) and epithelial cells (Misinzo et al., 2009) using immunofluorescence methods of detection.
When PCV2 enters the cytoplasm a replication process involving several cellular organelles is initiated. Our results suggest that the mitochondria may play a crucial role in the early stages of infection, similar to what has been described in lymph nodes of PMWS-affected animals (Rodríguez-Cariño and Segalés, 2009; Rodríguez-Cariño et al., 2010). In these previous invivo studies, VLPs were found to be closely associated with mitochondria, with PCV2 immunolabelling of both the inner and outer membranes. In the present in-vitro study, at 0 hpi the cells of the L35-derived clone showed ICIs adjacent to mitochondria and 6 h later the mitochondrial matrix contained VLPs. These organelles were successfully immunolabelled with the PCV2 antibody at 24 hpi and at subsequent time points. This finding suggests that PCV2 virions, once in the cytoplasm, enter the mitochondria or the nucleus, probably by endocytosis. Interestingly, previous ultrastructural studies of lymph nodes of pigs with PMWS showed macrophages with degenerate mitochondria containing VLPs (Rodríguez Cariño and Segalés, 2009). Moreover, the co-localization of PCV2 and mitochondrial antigens was demonstrated in this organelle (Rodríguez Cariño et al., 2010).
At 12 hpi, aggregates of icosahedral VLPs not surrounded by a membrane were observed in the nucleus. These aggregates were similar to those observed for PCV1 in PK15 cells (Stevenson et al.,1999). Nuclear localization of PCV2 has been previously described in PK15 cells using immunofluorescence methods (Cheung and Bolin, 2002; Meerts et al., 2005; Zhang et al., 2008), at 24 hpi. Therefore, it appears that the PCV2 replication in the L35- derived clone might be faster than in PK15 cells or, alternatively, that EM may be more sensitive than other detection methods such as immunofluorescence.
Morphological changes observed in successive hours post-inoculation indicate that PCV2 replication occurred in the nuclei of the L35-derived clone cells, where immature virions of varying shapes and sizes were observed. At the end of the nuclear phase of replication, clusters of VLPs were observed in proximity to protein networks, which may play a role in the final stages of virus assembly. Later (around 24 hpi), immature virions were apparently encapsidated at the periphery of the nuclear membrane and subsequently released into the cytoplasm by budding. These findings are in agreement with what has been described for immunofluorescence studies of PCV2 infection in PK15 culture cells (Meerts et al., 2005; Finsterbusch and Mankertz, 2009). Nuclear PCV2 DNA replication and encapsidation of the circular closed ssDNA occurs in the nucleus and not in cytoplasmic compartments (Finsterbusch and Mankertz, 2009), as has been described for other DNA viruses (Cullen, 2001; Finsterbusch and Mankertz, 2009). At 48 hpi, several ICIs with icosahedral VLPs arranged in paracrystalline arrays were observed, similar to those reported by Stevenson et al. (1999) in PCV1-infected PK15 cells. These immature virions were smaller than those detected in the nucleus. Such differences in VLP size are most likely due to molecular changes that affect the structural conformation of the virus particle, as has been suggested in the case of other viruses (Novoa et al., 2005b; Chichón et al., 2009).
Importantly, VFs were also observed within the cytoplasm, as has been described in histiocytes of pigs with PMWS (Rodríguez Cariño and Segalés, 2009). Unlike INIs, these cytoplasmic VFs were bound by double or triple membranes, suggesting the participation of other organelles in virus replication, transport and assembly as recognized in other viral infections (Novoa et al., 2005a). Therefore, it is highly likely that PCV2 may use the Golgi complex and RER for viral assembly and may approach the plasma membrane in a similar fashion to what has been described for Bunyamwera virus (Fontana et al., 2008). From 48 hpi onwards some cells showed clusters of VLPs budding from the cytoplasmic membrane. This cell release strategy has only previously been described for enveloped DNA viruses (Patient et al., 2009). Clusters of PCV2 particles may behave as a collective unit and move from organelle membranes to the cytoplasmic compartment in the final stage of the replication cycle. Finally, viral release requires the use of the cellular membrane for budding. Viral egress by exocytosis should occur in PCV2-infected cells, as this happens with other DNA viruses (Denesvre et al., 2007); however, the limited size of the PCV2 particles prevented their ultrastructural visualization. It is important to note that the number of dead cells increased in the last hours post-inoculation. The majority of these cells showed large clusters of virions arranged in paracrystalline arrays with a herringbone pattern. This may suggests that cell lysis is another route for the release of PCV2 from the cell, as has been described for a number of DNA viruses (Jiang et al., 2008).
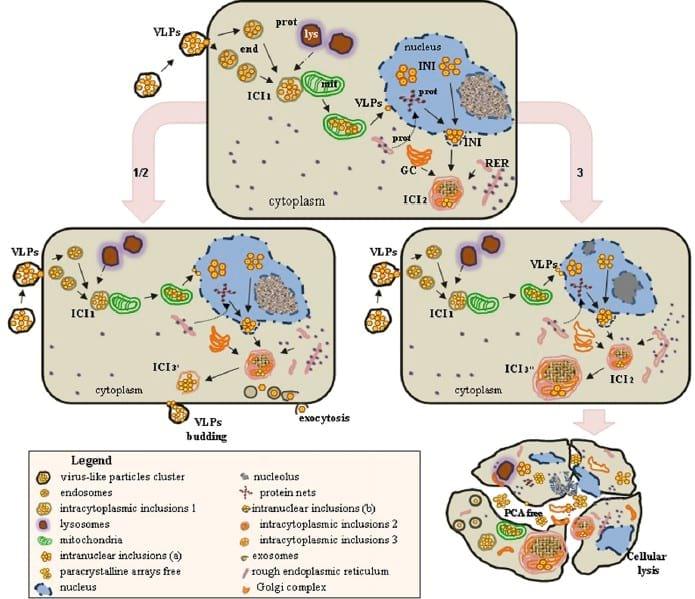
Fig. 7. Proposed scheme for PCV2 morphogenesis in the clone derived from the L35 lymphoblastoid cell line. An initial cytoplasmic phase (0-12 hpi) implies virion entrance into the cells by endocytosis (end) and arrangement into intracytoplasmic inclusions (ICI1). During this period, VLPs may attach and enter mitochondria (mit) and the nucleus. During the nuclear phase, different INIs are observed and are first located near heterochromatin and protein networks. At around 24 hpi, INIs arelocated near to the nuclear membrane; viral assembly and encapsidation appears to take place with the participation of undetermined nuclear elements and the nuclear membrane. A second cytoplasmic phase occurs with the recruitment and association of organelle membranes, including the Golgi complex and rough endoplasmic reticulum (RER).Asecond wave of ICIs appears (ICI2), which might evolve by different routes including: (1) conversion to a more simple conformation (ICI3a), which moves close to the cell membrane and VLPs as a unit and is released by budding, (2) alternatively, ICI2 might disaggregate and free VLPs may leave the cell by exocytosis, or (3) large and complex ICIs might be formed (ICI3b) with significant subcellular alterations (dilation of Golgi complexand RER) and, finally, cellular lysis to release immature and mature virions.
In summary, the present study has used conventional TEM and immunogold labelling to demonstrate new features of PCV2 morphogenesis. The findings, together with previously published information (Meerts et al., 2005; Misinzo et al., 2009), permitted us to propose a sequence of events that may occur during the PCV2 replication cycle (Fig. 7). It is important to point out the participation of mitochondria in the first cytoplasmic phase of the viral cycle, the ultrastructural confirmation of the important phase of nuclear replication, the progress of viral particles through the nuclear membrane and its participation in viral encapsidation. A second cytoplasmic phase occurs in association with organelle membranes. At the end of replication, mature virus particles may be released from cells by three routes: cell lysis, ICI budding and exocytosis. Finally, this study emphasizes the suitability of the L35-derived clone as an in-vitro model for studying PCV2- ost cell interactions.
Acknowledgments
The authors are grateful to personnel of the Servei de Microscopia from UAB (especially F. Cardoso and A. Dols-Pérez), for their excellent technical support, and Dr F. Rodríguez and Mr. J. Pujols from Centre de Recerca en Sanitat Animals (CReSA) for theirscientific advice. This work was partly funded by projects QLRT-PL-1999-0307 and 513928 from the Fifth and Sixth Framework Programmes of the European Commission, respectively, and the Consolider Ingenio 2010-PORCIVIR (Spanish Government). PhD studies of Mrs. C. Rodríguez Cariño are partially funded by a bursary from the Universidad Central de Venezuela.
References
Allan G, Krakowka S, Ellis J (2002) PCV2: ticking time bomb. Pig Progress, 18, 14e15.
Allan GM, McNeilly F, Cassidy JP, Reilly GA, Adair B et al. (1995) Pathogenesis of porcine circovirus; experimental infections of colostrum deprived piglets and examination of pig foetal material. Veterinary Microbiology, 44, 49e64.
Cheung AK, Bolin SR (2002) Kinetics of porcine circovirus type 2 replication. Archives of Virology, 147, 43e58.
Cullen BR (2001) Journey to the center of the cell. Cell, 105, 697e700.
Chicho´n FJ, Rodríguez MJ, Risco C, Fraile-Ramos A, Ferna´ ndez JJ et al. (2009) Membrane remodelling during vaccinia virus morphogenesis. Biology of the Cell, 101, 401e414.
Denesvre C, Blondeau C, Lemesle M, Le Vern Y, VautherotDet al. (2007) Morphogenesis of a highly replicative EGFPVP22 recombinant Marek´s disease virus in cell culture. Journal of Virology, 81, 12348e12359.
Ellis J, Hassard L, Clark E, Harding J, Allan G et al. (1998) Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Canadian Veterinary Journal, 39, 44e51.
Finsterbusch T, Mankertz A (2009) Porcine circoviruses e small but powerful. Virus Research, 143, 177e183.
Finsterbusch T, Steinfeldt T, Caliskan R, Mankertz A (2005) Analysis of the subcellular localization of the proteins Rep, Rep0 and Cap of porcine circovirus type 1. Virology, 343, 36e46.
Fontana J, Lopez-Montero N, Elliott RM, Fernandez JJ, Risco C (2008) The unique architecture of Bunyamwera virus factories around the Golgi complex. Cellular Microbiology, 10, 2012e2028.
Jiang H, White EJ, Go´mez-Manzano C, Fueyo J (2008) Adenovirus´s last trick: you say lysis, we say autophagy. Autophagy, 4, 118e120.
Kaeffer B, Bottreau E, Phan Thanh L, Olivier M, Salmon H (1990) Histocompatible miniature, boar model: selection of transformed cell lines of B and T lineages producing retrovirus. International Journal of Cancer, 46, 481e488.
Mankertz A, Caliskan R, Hattermann K, Hillenbrand B, Kurzendoerfer P et al. (2004) Molecular biology of porcine circovirus: analyses of gene expression and viral replication. Veterinary Microbiology, 98, 81e88.
McNair I, McNeilly F, Gilpin DF, Duffy C, Allan GM (2005) Growth of porcine circovirus 2 in lymphocyte cell lines. In: Proceedings of the European Society for Veterinary Virology, Belfast, p. 37.
McNeilly F, McNair I, Mackie DP, Meehan BM, Kennedy S et al. (2001) Production, characterisation and applications of monoclonal antibodies to porcine circovirus 2. Archives of Virology, 146, 909e922.
Meerts P, Misinzo G, McNeilly F, Nauwynck HJ (2005) Replication kinetics of different porcine circovirus 2 strains in PK-15 cells, fetal cardiomyocytes and macrophages. Archives of Virology, 150, 427e441.
Misinzo G, Delputte PL, Lefebvre DJ, Nauwynck HJ (2009) Porcine circovirus 2 infection of epithelial cells is clathrin-, caveolae- and dynamin-independent, actin and Rho-GTPase-mediated, and enhanced by cholesterol depletion. Virus Research, 139, 1e9.
Misinzo G, Meerts P, Bublot M, Mast J, Weingartl HM et al. (2005) Binding and entry characteristics of porcine circovirus 2 in cells of the porcine monocytic line 3D4/ 31. Journal of General Virology, 86, 2057e2068.
Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H et al. (2005a) Virus factories: associations of cell organelles for viral replication and morphogenesis. Biology of the Cell, 97, 147e172.
Novoa RR, Calderita G, Cabezas P, Elliott RM, Risco C (2005b) Key Golgi factors for structural and functional maturation of Bunyamwera virus. Journal of Virology, 79, 10852e10863.
Patient R, Hourioux C, Roingeard P (2009) Morphogenesis of hepatitis B virus and its subviral envelope particles. Cellular Microbiology, 11, 1561e1570.
Rodríguez-Cariño, Sánchez-Chardi A, Segalés J (2010) Subcellular immunolocalization of porcine circovirus type 2 (PCV2) in lymph nodes from pigs with postweaning multisystemic wasting syndrome (PMWS). Journal of Comparative Pathology, 142, 291e299.
Rodríguez Cariño C, Segalés J (2009) Ultrastructural findings in lymph nodes from pigs suffering from naturally occurring postweaning multisystemic wasting syndrome. Veterinary Pathology, 46, 729e735.
Segalés J, Allan GM, Domingo M (2005) Porcine circovirus diseases. Animal Health Research Reviews, 6, 119e142.
Stevenson GW, Kiupel M, Mittal SK, Kanitz CL (1999) Ultrastructure of porcine circovirus in persistently infected PK-15 cells. Veterinary Pathology, 36, 368e378.
Tischer I, Rasch R, Tochtermann G (1974) Characterization of papovavirus-like and picornavirus-like particles in permanent pig kidney cell lines. Zentralblatt fu¨r Bakteriologie, 226, 153e167.
Toma´ s A, Fernandes LT, Valero O, Segalés J (2008) A meta-analysis on experimental infections with porcine circovirus type 2 (PCV2). Veterinary Microbiology, 132, 260e273.
Vincent IE, Balmelli C, Meehan B, Allan G, Summerfield A (2007) Silencing of natural interferon producing cell activation by porcine circovirus type 2 DNA. Immunology, 120, 47e56.
Zhang X, Ma G, Li Y, Jiang X, He J et al. (2008) Characterization of monoclonal antibody against replicationassociated protein of porcine circovirus. DNA Cell Biology, 28, 23e29.
This article was originally published by Elsevier in 2010. Engormix.com thanks for this contribution.