1 Introduction
The Fusarium genera include a diverse group of phytopathogenic fungi that are dispersed around the world. Inside this genera are several species that are able to produce mycotoxins, including Fusarium graminearum, which is a pathogen of maize and wheat. In wheat and in other winter cereals, it causes Fusarium head blight (FHB), or scab, which is the main disease of these cereals in the Southern region of Brazil (PARRY; JENKINSON; McLEOD, 1995; McMULLEN; JONES; GALLENBERG, 1997; GALE, 2003). Epidemics of this disease cause economic losses due to the reduced grain quality and due to the toxic effect of the produced mycotoxins.
Mycotoxins are chemical compounds resulting from the secondary metabolism of filamentous fungi, which in small concentration can cause chronic or acute diseases in vertebrate animals (RICHARD, 2007). Among the mycotoxins produced by F. graminearum are deoxynivalenol (DON), nivalenol (NIV), and zearalenone (ZEA). Isolates of F. graminearum that produce DON may also produce two acetylated derivatives of DON, 3-acetyl-deoxynivalenol (3-ADON) and 15-acetyldeoxynivalenol (15-ADON) (MILLER et al., 1991). DON and NIV are trichothecenes and ZEA is a non steroidal estrogen. Trichothecenes are immunosuppressive and can cause renal problems in animals (GERALDO; TESSMANN; KEMMELMEIER, 2006; RICHARD, 2007) and zearalenone has estrogenic effects (MIROCHA et al., 1978).
DON and ZEA detection analysis is performed by chromatographic methods, which are laborious, time-consuming, expensive, require well trained personal, and use several toxic reagents. The F. graminearum detection in culture or in infected grains is carried out, traditionally, by micro and macro morphologic analysis. These processes are also time-consuming, besides requiring well trained personal, and are not precise when the distinction among similar species is difficult.
In spite of the frequent use of molecular methods, more sensitive and faster, for the detection and identification of bacteria in food, few molecular methods have been developed for detection of fungi in those substrates. However, molecular methods are employed for the specific identification of Fusarium species in culture or in affected grains. In this regard, the polymerase chain reaction (PCR) has been described for the detection and specific identification of F. graminearum (SCHILING; MÖLLER; GEIGER, 1996; NIESSEN; VOGEL, 1997; NICHOLSON et al., 1998; TAN; NIESSEN, 2003; JURADO et al., 2005; NIESSEN, 2007; BIAZIO et al., 2008). PCR assays have also been developed to rapidly assess trichothecene mycotoxin genotypes of F. graminearum. Such assays rely on the amplification of portions of genes that code for key enzymes involved in trichothecene biosynthesis (LEE et al., 2001; WARD et al., 2002; CHANDLER et al., 2003; JENNINGS et al., 2004; QUARTA et al., 2006). These assays are attractive because they are rapid, relatively inexpensive, and may be used to accurately predict the types of trichothecene mycotoxins produced by the isolates in culture (WARD et al., 2002).
Several studies report a high correlation between fungal infection and occurrence of mycotoxins (GONZÁLES et al., 1999; LORI et al., 2003). Therefore, the molecular detection of F. graminearum with trichothecene genotype in foodstuff could represent a potential indicative of DON and ZEA contamination. Bulgur wheat is a cereal food made from wheat grains. This cereal is used in the Mediterranean and Arabic cuisine and is greatly appreciated in Brazil. Therefore, the objective of the present study was to develop a PCR assay to analyze the presence of F. graminearum in bulgur wheat commercialized in the city of Maringá, State of Paraná, Brazil. Two approaches were used, one tested the possibility to detect the presence of F. graminearum DNA directly in the food, and the other tested the possibility to isolate and molecularly identify F. graminearum in food cultures.
2 Material and methods
2.1 Samples
The commercial sample (500 g) used for contamination with the F. graminearum homogenized culture was collected by the Paraná State Health Agency in the second semester of 2007. The sample was grounded, homogenized, and stored at –20 °C until analyses. For the survey analyses, 42 commercial samples, individually packaged in plastic bags (500 g each), regularly commercialized in stores were acquired in the period from July/2008 to June/2009 in the city of Maringá, State of Paraná, which is located in Southern Brazil, the main wheat producer region in Brazil. These samples belonged to 11 different commercial brands and from different batches. All of them were analyzed within the expiration dates. These samples were stored at the room temperature.
2.2 Sample contamination
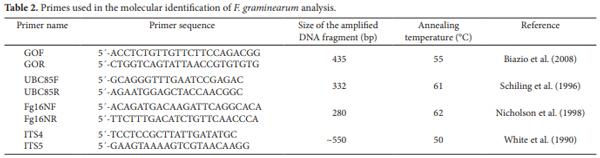
To test the smallest amount of fungal contamination on bulgur wheat that could be detected using DNA extraction and PCR with specific primers, a sample was contaminated with a homogenized F. graminearum liquid culture. The strain of F. graminearum used as a reference was kindly provided by Dr. Carlos Kemmelmeier (Universidade Estadual de Maringá, PR, Brazil). This strain is the producer of the enzyme galactose oxidase (COOPER et al., 1959; AVIGAD et al., 1962) and was deposited in the NRRL collection by the identifiers and received the number 2903. The homogenized liquid culture was obtained as follows. A fragment of one cm3 of a fresh culture in inclined potato dextrose agar (PDA) was transferred to a 125 mL Erlenmeyer flask containing 25 mL of the liquid medium described by Biazio et al. (2008). This medium was prepared in three separated solutions: solution A: 62 mM Na2 HPO4 , 62 mM KH2 PO4 , 13 mM (NH4 )NO3 , 15 mM (NH4 )2 SO4 , 15 mM NaOH, 14 mM KOH, 0.1% yeast extract, pH 7.0; solution B: 1.6 mM MgSO4 •7H2 O, 11.8 µM MnSO4 •H2 O, 10 µM CuSO4 •5H2 O; and solution C: 1% glucose. Solutions A and B were autoclaved at 121 °C for 20 minutes, and solution C was autoclaved without pressure for the same time. Before use, 22.5 mL of the solution A, 0.5 mL of the solution B, and 2.0 mL of the solution C were combined aseptically. The indicated concentrations are those in the final medium. After the inoculation, the flasks were incubated for four days at 25 °C under agitation (100 rpm) and the obtained culture was homogenized by passage firstly through a sterile steel sieve and secondly by using a18G sterile needle attached to a sterile syringe. This homogenized culture solution was mixed with two grams of the bulgur wheat sample in the following proportions: 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 20, and 40% (volume/mass).
2.3 F. graminearum survey analysis
One hundred miligrams of each bulgur wheat sample were spread over a 10 cm Petri dish containing PDA supplemented with 50 µg.mL–1 of chloranfenicol, in triplicates. The dishes were incubated for four to seven days. The incubations (for growth and development of microorganisms) were performed in an incubator at 25 °C with a photoperiod of 12 hours. This methodology was repeated twice, and each sample was analyzed in a total of six dishes. Fungi colonies that pigmented the PDA with red color were transferred to tubes with inclined PDA, which were incubated for five to seven days. For the monosporic isolation, a fragment of approximately one cm3 of the obtained culture in the inclined PDA was smashed and agitated in 10 mL of distilled water. One hundred microliters of the obtained suspension were spread over the surface of 1.5% water-agar in a 10-cm Petri dish. After 18 to 24 hour incubation, a fragment of the medium containing a germinating spore or a hypha fragment was removed and transferred to an inclined PDA tube. This tube was incubated for five to seven days, and the culture characteristics were observed.
2.4 DNA extraction
The genomic DNA was extracted following the method described by Koenig, Ploetz and Kistler (1997) from 0.2 g of the contaminated and non contaminated samples of bulgur wheat or from the mycelia of each obtained isolated in the bulgur wheat culture technique (Table 1). The mycelia of each monosporic isolate were obtained by filtration in sterile gauze of a five-day culture. This culture was carried out in a 125-mL flask containing 25 mL of liquid potato dextrose medium that was inoculated with two mL of a suspension produced either by chopping and shaking a one cm3 piece of a fresh culture in an inclined PDA tube in 5 mL of sterile distilled water or by scraping a fresh culture in a PDA Petri dish with 5 mL of sterile distilled water. For the DNA extraction, the bulgur wheat or the mycelia were macerated in liquid nitrogen, and transferred to microcentrifuge tubes. The extraction buffer was added in the proportion of 700 μL per each 300 μL of macerated material. The extraction buffer contained, in the proportion of 1.0/1.0/0.4, nuclear lyses buffer (0.2 M Tris, pH 7.5; 50 mM EDTA; 2% (w/v) cetyltrimethylammonium bromide; pH 7.5), DNA isolation buffer (0.35 M sorbitol; 0.1 M Tris, pH 7.5; 5 mM EDTA; pH 7.5), and 5% Sarkosyl. The extraction buffer was combined right before use and was then added with 3.8 mg.mL–1 of sodium bisulfite. The tubes were incubated in a dry bath at 65 °C for 60 minutes. After that, 500 µL of a mixture of chloroform:isoamylic alcohol (24:1) were added. The tubes were centrifuged at room temperature (12.000 g, 10 minutes), and the supernatant fraction was transferred to clean tubes. Samples were treated with five μl of RNAse A (20 mg.mL–1), for 30 minutes, at 37 °C, and next with 5 µL of proteinase K (20 mg.mL–1), for 30 minutes, at 56 °C. The DNA was then precipitated with an equal volume of isopropanol and was incubated at –20 °C overnight. The precipitated DNA was collected by centrifugation at room temperature (12.000 g, 10 minutes), and the DNA pellet was washed three times with cold 70% ethanol. The final DNA pellet was dried at room temperature and resuspended with 50 µL of TE buffer (10 mM Tris, pH 8.0; 1 mM EDTA). The whole process was carried out under sterile conditions. The DNA was quantified in a spectrophotometer at 260 nm. The DNA final concentration was adjusted to 100 ng.µL–1 in TE buffer, and the DNA was kept frozen at –20 °C.
2.5 PCR analysis
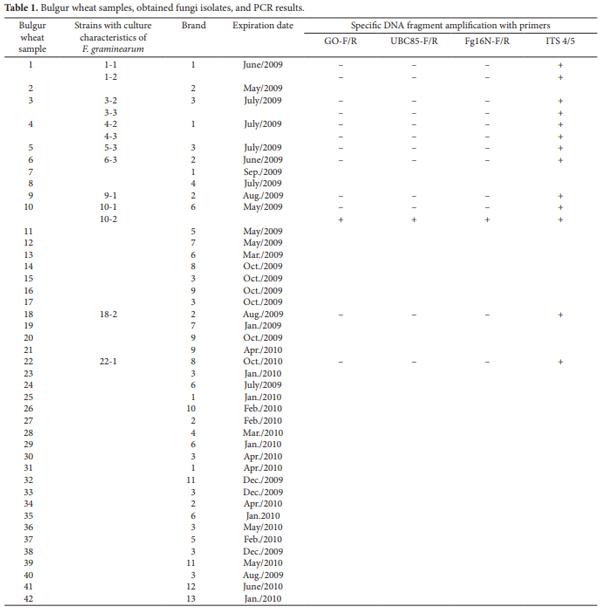
The primer pair used to test the smallest amount of fungal contamination in bulgur wheat that could be detected using PCR was the GO primers (Table 2), described by Biazio et al. (2008). The primers used in the PCR reactions with the DNA extracted from the isolates obtained from culturing the bulgur wheat were the ones described for F. graminearum specific identification: UBC85 (SCHILING; MÖLLER; GEIGER, 1996), Fg16N (NICHOLSON et al., 1998), and GO (BIAZIO et al., 2008) (Table 2). All genomic DNAs used in this study were tested for PCR amplification suitability using the ITS4 and ITS5 primers targeted to the ITS-5.8 S rRNA region (WHITE et al., 1990). A DNA fragment of approximately 550 bp was amplified from all genomic DNAs using the ITS4 and ITS5 primers indicating that they were suitable for amplification. The amplification reactions were performed in a termocycler Techne TC-312 (England) in PCR tubes containing 25 µL of the following reaction mixture: 1 X enzyme buffer; 1.5 mM MgCl2 , 1.5 U of Platinum Taq DNA polymerase (Invitrogen); 0.2 mM of each dNTP (Invitrogen); 25 pmol of each primer (forward and reverse), and 20-400 ng of the DNA sample. The PCR reaction consisted of 25 cycles of 1 minute and 30 seconds at 94 °C, 1 minute and 30 seconds at the annealing temperature listed in Table 2, and 2 minutes at 72 °C. Prior to the cycles, the samples were heated for 5 minute at 94 °C, and after the cycles the samples were incubated for 10 minutes at 72 °C and frozen at –20 °C until use. Positive control was the DNA from the isolate F. graminearum (NRRL 2903). Negative controls (no DNA template) were used in each experiment to test for the presence of DNA contamination of reagents and reaction mixtures. Ten microliters of the PCR reaction were analyzed in a 1.5% agarose gel containing ethidium bromide (0.25 µg.mL–1). The PCR products were visualized and photographed under UV light.
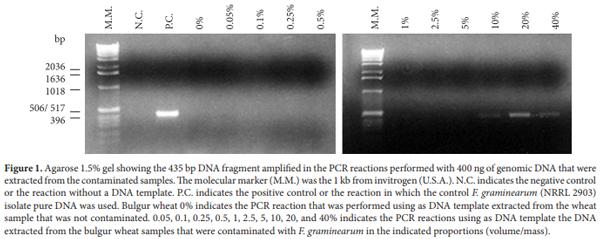
2.6 Trichothecenes genotyping of the F. graminearum isolate
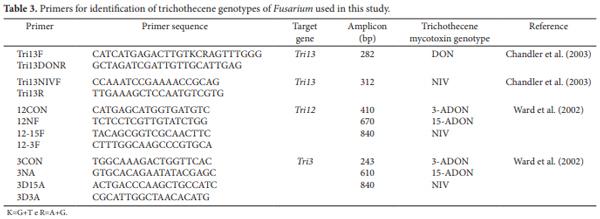
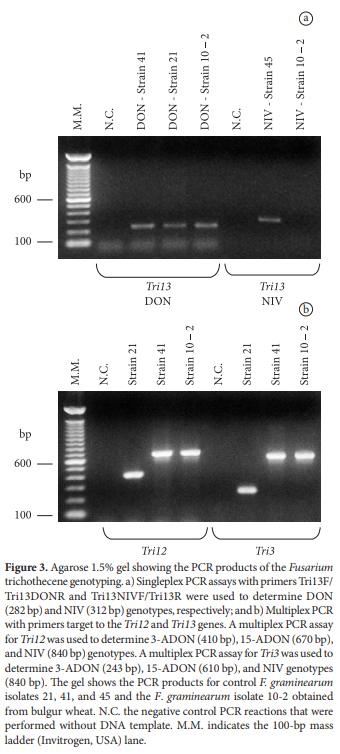
DON and NIV genotypes were identified using an essay to amplify portions of the Tri13 gene (CHANDLER et al., 2003). DON, 3-ADON, 15-ADON, and NIV genotypes were identified using a multiplex reaction to identify the Tri12 and Tri3 genes (WARD et al., 2002). With the Tri12 primer set, PCR products of 410, 670, and 840 bp are produced for 3-ADON, 15-ADON, and NIV genotypes, respectively (WARD et al., 2002). With the Tri3 primer set, PCR products of 243, 610, and 840 bp are produced for 3-ADON, 15- ADON, and NIV genotypes, respectively (WARD et al., 2002). All information on primer sequences and amplicon sizes for the respective trichothecene mycotoxin genotypes is summarized in Table 3.
PCR assays were conducted using 400 ng of fungal DNA in a total volume of 25 µL containing 1.5 mM MgCl2 , 2U of Platinum Taq DNA polymerase (Invitrogen, USA), and 0.2 mM of each dNTPs. Positive controls consisted of the DNA isolated from F. graminearum strains 21, 41, and 45 that were obtained from wheat and were described previously (ANGELOTTI et al., 2006; GERALDO; TESSMANN; KEMMELMEIER, 2005; BIAZIO et al., 2008). The DNA of these strains was extracted as described above. F. graminearum strain 21 has a genotype of DON and 3-ADON production, F. graminearum strain 41 has a genotype of DON and 15-ADON production, and F. graminearum strain 45 has a genotype of NIV production. PCR amplification of Tri13 was performed as described above with an annealing temperature of 60 °C. PCR amplification of Tri12 and Tri3 consisted of an initial step at 94 °C for 10 minutes, followed by two cycles of 94 °C for 30 seconds, 59 °C for 30 seconds, and 72 °C for 30 seconds. The annealing temperature was stepped down every two cycles to 58, 56, 54, 53, 52, and 51 °C, and next 50 °C for 21 cycles, with a final step at 72 °C for 10 minutes. Ten microliters of the PCR reaction were analyzed in an agarose gel containing ethidium bromide (0.25 µg.mL–1). The PCR products were visualized and photographed under UV light.
3 Results and discussion
The information about the presence of fungi in grains is of extreme importance for the evaluation of mycotoxin contamination risk in the derived food, mainly in wheat, which is the basic cereal source for several communities in Brazil and around the world. In spite of the numerous studies about the presence of mycotoxins in foods, literature about fungi presence is scarce.
The results obtained when a bulgur wheat sample was contaminated with F. graminearum mycelia and the DNA was extracted and used for PCR reactions (Figure 1) show that the methodology used was effective in extracting and amplifying F. graminearum DNA from a bulgur wheat sample that was contaminated at the concentration of 10, 20, and 40% (volume/mass). This indicates a small sensitivity of the standardized methodology. The used genomic DNA extraction method does extract DNA from the fungi and from the cereal. Since the cereal was always in higher amount, it is probable that the majority of the DNA added to the PCR reaction was from the cereal and not from the fungi. Since the primers used had a high sensitivity (BIAZIO et al., 2008), it is probable that the obtained negative results are due to a very small amount of the fungi genomic DNA present in the poorly contaminated samples or due to PCR reaction inhibition caused by the cereal DNA. PCR reactions performed with higher and smaller amounts of genomic DNA extracted from the contaminated cereal did not result in increased sensitivity (not shown). Commercial samples in store shelves have low probability to be contaminated with a high amount of fungi mycelia because of the cereal low level of humidity. Hence, the developed methodology would not be appropriate to test the presence of mycotoxigenic F. graminerum in commercial samples of bulgur wheat. However, it could be useful in field samples that are more susceptible to contamination. Accordingly, Jurado et al. (2006) found a high amount of Fusarium verticillioides using a PCR reaction with genomic DNA extracted directly from mycotoxins contaminated maize seeds. In addition, Kulik et al. (2007) also observed the presence of Fusarium spp. in a high number of wheat samples that were collected directly from the field.
The majority of studies that identify the presence of fungi in food are based on the cereal culture in selective media, in the isolation of colonies with compatible culture characteristics, and in the isolates morphological analysis (TONON et al., 1997; PARK et al., 2005). When molecular biology techniques such as PCR are used, they employ as template the obtained isolates genomic DNA and not the genomic DNA obtained directly from the food (BLUHM et al., 2002). Therefore, this methodology was also employed in this study
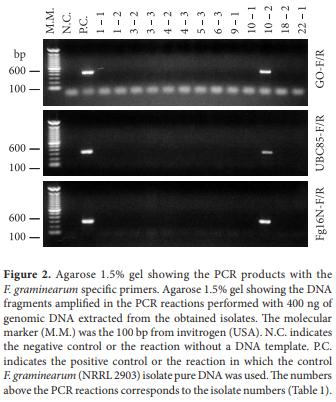
Forty two bulgur wheat samples were cultured (Table 1). Thirteen monosporic isolates that produced a red pigment in PDA, a culture characteristic of F. graminearum, were obtained from nine of the 42 bulgur wheat samples. From these thirteen isolates, only one was molecularly identified as F. graminearum in PCR reactions with the GO, UBC85, and FG16N primers (Figure 2). This indicates a low contamination of the samples by F. graminearum (one in forty two or 2.38%). It should be taken into consideration that the analyzed samples were commercial type samples that had passed quality control tests and that were processed for packing. If this methodology had been used with food samples derived directly from the field, it could have resulted in a higher number of contaminated samples and could be used as a quality control method before processing for packing. The presence of F. graminearum in commercial bulgur wheat indicates, therefore, that in the presence of moisture this fungus could proliferate and produce mycotoxins. Thus, care should be taken in controlling the humidity of this product in store shelves.
The obtained F. graminearum isolate (strain 10-2) was tested for its trichothene genotype. This isolate was identified to have a DON (Figure 3a) and 15-ADON (Figure 3b) genotype, which indicates that it has a potential to produce these mycotoxins. This data is in agreement with reports about the predominance of trichothecene genotypes for DON and 15-ADON in F. graminearum isolates from Argentina and Southern Brazil (RAMIREZ et al., 2006; SCOZ et al., 2009).
The methodology recommended by the International Commission on Food Mycology (ICFM), a commission of the International Union of Microbiological Societies (IUMS), for isolation, enumeration, and identification of fungi from foods consists of techniques based on viable counting (dilution plating and direct plating) for detecting and quantifying fungal growth in foods (PITT; HOCKING, 2009). The direct plating is the preferred method for particulate foods such as grains and nuts. In this method, food particles are disinfected and placed directly on solidified agar media. Two types of media have been used for the enumeration and identification of Fusarium isolates: PDA and dichloran chloramphenicol peptone agar (DCPA) (PITT; HOCKING, 2009). Fungi grown in plates are submitted to monosporic isolation and analyzed in microculture in DCPA and carnation leaf agar (CLA) for identification. The identification of Fusarium is based on the morphology of the macro and microconidia and in the colony characteristics and color from cultures grown on PDA (PITT; HOCKING, 2009). When comparing the official methodology and the methodology used in this study, the initial steps are similar but the identification step is different; the former uses traditional morphology and the latter uses a molecular method. Considering time, work involved, sensitivity, and costs, both methodologies are time-consuming, laborious, sensitive, and have approximately the same cost. The molecular method could be considered more expensive because of the high cost necessary for its establishment. The main advantage of the proposed molecular methodology regards the specific species identification that can be done without the need of morphological identification of the isolates, which is error-prone. In addition, the morphological identification gives no indication of the mycotoxigenic profile of the obtained isolates, which can be easily obtained with the molecular method.
4 Conclusions
In conclusion, a methodology to identify the presence of mycotoxigenic F. graminearum in food was developed in this study. This methodology consisted in food culturing and isolation of fungi with culture characteristics of F. graminearum. After that, the isolates were molecularly identified by PCR with specific primers and had the genotype for trichothecene production determined by PCR with primers directed to genes of enzymes involved in mycotoxins synthesis. This methodology is of easy application and could be used in surveys of food and cereal contamination.
This article was originally published in Ciênc. Tecnol. Aliment., Campinas, 32(1): 201-208, jan.-mar. 2012. DDOI: http://dx.doi.org/10.1590/S0101-20612012005000027. This is an Open Access article licensed under a Creative Commons Attribution License. References
ANGELOTTI, F. et al. Caracterização morfológica e identificação molecular de isolados de Fusarium graminearum associados à giberela do trigo e triticale no sul do Brasil. Summa Phytopathologica, v. 32, n, 2, p. 177-179, 2006. http://dx.doi.org/10.1590/S0100-54052006000200013
AVIGAD, G. et al. The D-galactose oxidase of Polyporus circinatus. Journal of Biological Chemistry, v. 237, n. 9, p. 2736-2743, 1962. PMid:13863403
BIAZIO, G. R. et al. A new PCR approach for the identification of Fusarium graminearum. Brazilian Journal of Microbiology, v. 39, n. 3, p. 554-560, 2008. http://dx.doi.org/10.1590/S1517- 83822008000300028
BLUHM, B. H. et al. Multiplex polymerase chain reaction assay for the differential detection of trichothecene- and fumonisin-producing species of Fusarium in cornmeal. Journal of Food Protection, v. 65, n. 12, p. 1955-1961, 2002. PMid:12495016
CHANDLER, E. A. et al. Development of PCR assays to Tri7 and Tri13 trichothecene biosynthetic genes, and characterisation of chemotypes of Fusarium graminearum, Fusarium culmorum and Fusarium cerealis. Physiological and Molecular Plant Pathology, v. 62, n. 6, p. 355-367, 2003. http://dx.doi.org/10.1016/S0885- 5765(03)00092-4
COOPER, J. A. D. et al. Galactose oxidase from Polyporus circinatus, Fr. Journal of Biological Chemistry, v. 234, n. 3, p. 445-448, 1959. PMid:13641238
GALE, L. R. Population biology of Fusarium species causing head blight of grain crops, In: LEONARD, K. J.; BUSHNELL, W. R. (Ed.). Fusarium Head Blight of Wheat and Barley. Saint Paul: APS Press, 2003. cap. 5, p. 120-143.
GERALDO, M. R. F.; TESSMANN, D. J., KEMMELMEIER, C. Production of mycotoxins by Fusarium graminearum isolated from small cereals (wheat, triticale and barley) affected with scab disease in southern Brazil. Brazilian Journal of Microbiology, v. 37, n. 1, p. 58-63, 2006. http://dx.doi.org/10.1590/S1517- 83822006000100011
GONZÁLEZ, H. H. L. et al. Relationship betwenn Fusarium graminearum and Alternaria alternata contamination and deoxynivalenol occurrence on Argentina durum wheat. Mycopathology, v. 144, n. 2, p. 97-102, 1999. http://dx.doi.org/10.1023/A:1007020822134
JENNINGS, P. et al. Determination of deoxynivalenol- and nivalenolproducing chemotypes of Fusarium graminearum isolated from wheat crops in England and Wales. Plant Pathology, v. 53, n. 5, p. 643-652, 2004. http://dx.doi.org/10.1111/j.0032- 0862.2004.01061.x
JURADO, A. et al. PCR detection assays for the trichotheceneproducing species Fusarium graminearum, Fusarium culmorum, Fusarium poae, Fusarium equiseti and Fusarium sporotrichioides. Systematic and Applied Microbiology, v. 28, n. 6, p. 562-568, 2005. PMid:16104354. http://dx.doi.org/10.1016/j.syapm.2005.02.003
JURADO, A. et al. PCR-based strategy to detect contamination with mycotoxigenic Fusarium species in maize. Systematic and Applied Microbiology, v. 29, n. 8, p. 681-689, 2006. http://dx.doi. org/10.1016/j.syapm.2006.01.014
KOENIG, R. L.; PLOETZ, R. C.; KISTLER, H. C. Fusarium oxysporum f.sp. cubense consists of a small number of divergent and globally distributed clonal lineages. Phytopathology, v. 87, n. 9, p. 915-923, 1997. PMid:18945062. http://dx.doi.org/10.1094/ PHYTO.1997.87.9.915
KULIK, T. et al. PCR approach based on the esyn1 gene for the detection of potential enniatin-producing Fusarium species. International Journal of Food Microbiology, v. 116, n. 3, p. 319-324, 2007. PMid:17391792. http://dx.doi.org/10.1016/j. ijfoodmicro.2007.02.003
LEE, T. et al. Identification of deoxynivalenol- and nivalenolproducing chemotypes of Gibberella zeae by using PCR. Applied and Environmental Microbiology, v. 67, n. 7, p. 2966-2972, 2001. PMid:11425709 PMCid:92968. http://dx.doi.org/10.1128/ AEM.67.7.2966-2972.2001
LORI, G. A. et al. Fusarium graminearum and deoxynivalenol contamination in the durum wheat area of Argentina. Microbiological Research, v. 158, n. 1, p. 29-35, 2003. PMid:12608577. http://dx.doi. org/10.1078/0944-5013-00173
McMULLEN, M.; JONES, R.; GALLENBERG, D. Scab of wheat and barley: a re-emerging disease of devastating impact. Plant Disease, v. 81, n. 12, p. 1340-1348, 1997. http://dx.doi.org/10.1094/ PDIS.1997.81.12.1340
MILLER, J. D. et al. Trichothecene chemotype of three Fusarium species. Mycologia, v. 83, n. 2, p. 121-130, 1991. http://dx.doi. org/10.2307/3759927
MIROCHA, C. J. et al. Uterotropic activity of cis and trans isomers of zearalenone and zearalenol. Applied and Environmental Microbiology, v. 35, n. 5, p. 986-987, 1978. PMid:655715 PMCid:242968
NICHOLSON, P. et al. Detection and quantification of Fusarium culmorum and Fusarium graminearum in cereals using PCR assays. Physiological and Molecular Plant Pathology, v. 53, n. 1, p. 17–37, 1998. http://dx.doi.org/10.1006/pmpp.1998.0170
NIESSEN, L. PCR-based diagnosis and quantification of mycotoxin producing fungi. International Journal of Food Microbiology, v. 119, n. 1-2, p. 38-46, 2007. PMid:17804102. http://dx.doi. org/10.1016/j.ijfoodmicro.2007.07.023
NIESSEN, M. L.; VOGEL, R. F. Specific identification of Fusarium graminearum by PCR with gaoA targeted primers. Systematic and Applied Microbiology, v. 20, n. 1, p. 111-113, 1997. http://dx.doi. org/10.1016/S0723-2020(97)80055-0
PARK, J. W. et al. Fungal mycoflora and mycotoxins in Korean polished rice destined for humans. International Journal of Food Microbiology, v. 103, n. 3, p. 305-314, 2005. PMid:16099315. http:// dx.doi.org/10.1016/j.ijfoodmicro.2005.02.001
PARRY, D. W.; JENKINSON, P.; McLEOD, L. Fusarium ear blight (scab) in small grain cereals-A review. Plant Pathology, v. 44, n. 2, p. 207-238, 1995. http://dx.doi.org/10.1111/j.1365-3059.1995.tb02773.x
PITT, J. I.; HOCKING, A. D. Fungi and food spoilage. 3nd ed. New York: Springer, 2009. http://dx.doi.org/10.1007/978-0-387-92207-2
QUARTA, A. et al. Multiplex PCR assay for the identification of nivalenol, 3- and 15-acetyl-deoxynivalenol chemotypes in Fusarium. FEMS Microbiology Letters, v. 259, n. 1, p. 7-13, 2006. PMid:16684095. http://dx.doi.org/10.1111/j.1574-6968.2006.00235.x
RAMIREZ, M. L. et al. Vegetative compatibility and mycotoxin chemotypes among Fusarium graminearum (Gibberella zeae) isolates from wheat in Argentina. European Journal of Plant Pathology, v. 115, n. 2, p. 139-148, 2006. http://dx.doi.org/10.1007/ s10658-006-0009-1
RICHARD, J. L. Some major mycotoxins and their mycotoxicoses - An overview. International Journal of Food Microbiology, v. 119, n. 1-2, p. 3-10, 2007. PMid:17719115. http://dx.doi.org/10.1016/j. ijfoodmicro.2007.07.019
SCHILING, A. G.; MÖLLER, E. M.; GEIGER, H. H. Polymerase Chain Reaction-Based Assays for Species-Specific Detection of Fusarium culmorum, F. graminearum, and F. avenaceum. Phytopathology, v. 86, n. 5, p. 515-522, 1996. http://dx.doi.org/10.1094/Phyto-86-515
SCOZ, L. B. et al. Trichothecene mycotoxin genotypes of Fusarium graminearum sensu stricto and Fusarium meridionale in wheat from southern Brazil. Plant Pathology, v. 58, n. 2, p. 344-351, 2009. http:// dx.doi.org/10.1111/j.1365-3059.2008.01949.x
TAN, M.-K; NIESSEN, L. M. Analysis of rDNA ITS sequences to determine genetic relationship among, and provide a basis for simplified diagnosis of, Fusarium species causing crown rot and head blight of cereals. Mycological Research, v. 107, n. 7, p. 811-821, 2003. PMid:12967208. http://dx.doi.org/10.1017/S0953756203008013
TONON, S. A. et al. Mycoflora of paddy and milled rice produced in the region of Northeastern Argentina and Southern Paraguay. International Journal of Food Microbiology, v. 37, n. 2-3, p. 231-235, 1997. http://dx.doi.org/10.1016/S0168-1605(97)00066-4
WARD, T. J. et al. Ancestral polymorphism and adaptative evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proceedings of the National Academy of Sciences, USA, v. 99, n. 14, p. 9278-9283, 2002. PMid:12080147 PMCid:123131. http://dx.doi.org/10.1073/pnas.142307199
WHITE, T. J. et al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: INNIS, M. A. et al. (Ed.). PCR Protocols, a Guide to Methods and Applications. San Diego: Academic Press, 1990. cap. 38, p. 315-322.







.jpg&w=3840&q=75)