Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens
Background: Clostridium perfringens causes a range of diseases in animals and humans including necrotic enteritis in chickens and food poisoning and gas gangrene in humans. Necrotic enteritis is of concern in commercial chicken production due to the cost of the implementation of infection control measures and to productivity losses. This study has focused on the genomic analysis of a range of chicken-derived C. perfringens isolates, from around the world and from different years. The genomes were sequenced and compared with 20 genomes available from public databases, which were from a diverse collection of isolates from chickens, other animals, and humans. We used a distance based phylogeny that was constructed based on gene content rather than sequence identity. Similarity between strains was defined as the number of genes that they have in common divided by their total number of genes. In this type of phylogenetic analysis, evolutionary distance can be interpreted in terms of evolutionary events such as acquisition and loss of genes, whereas the underlying properties (the gene content) can be interpreted in terms of function. We also compared these methods to the sequence-based phylogeny of the core genome.
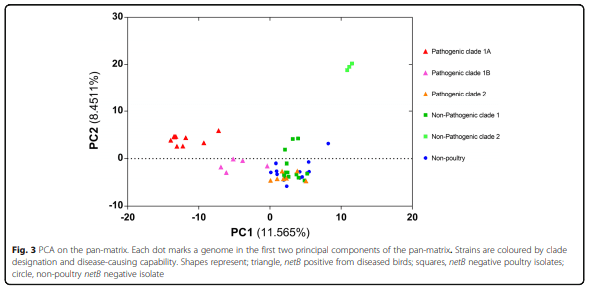
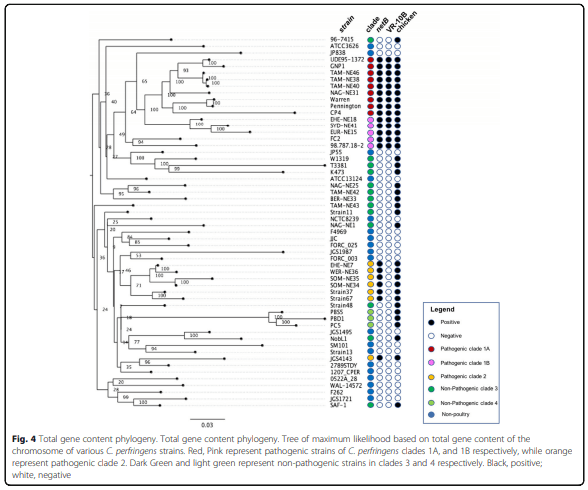
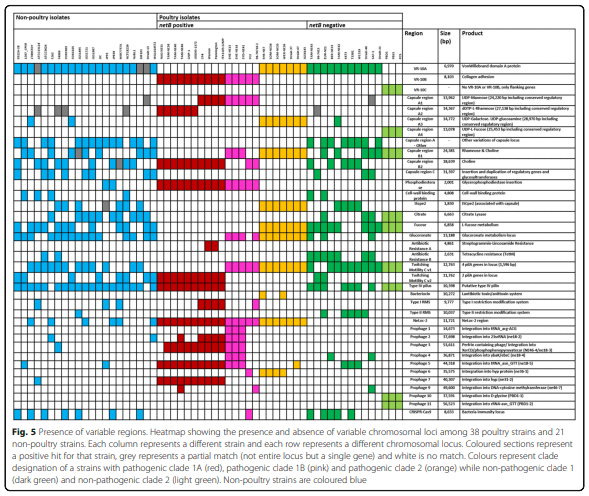
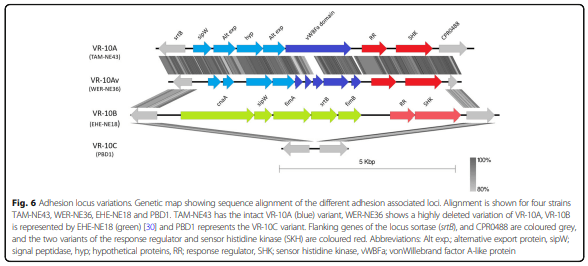
Results: Distinct pathogenic clades of necrotic enteritis-causing C. perfringens were identified. They were characterised by variable regions encoded on the chromosome, with predicted roles in capsule production, adhesion, inhibition of related strains, phage integration, and metabolism. Some strains have almost identical genomes, even though they were isolated from different geographic regions at various times, while other highly distant genomes appear to result in similar outcomes with regard to virulence and pathogenesis.
Conclusions: The high level of diversity in chicken isolates suggests there is no reliable factor that defines a chicken strain of C. perfringens, however, disease-causing strains can be defined by the presence of netB-encoding plasmids. This study reveals that horizontal gene transfer appears to play a significant role in genetic variation of the C. perfringens chromosome as well as the plasmid content within strains.
Keywords: Clostridium perfringens, Necrotic enteritis, Capsule, Adhesion, Prophage, Genome, Pangenome.

1. Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, et al. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014;9:361–77.
2. Li J, Adams V, Bannam TL, Miyamoto K, Garcia JP, Uzal FA, et al. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev. 2013;77:208–33.
3. Parreira VR, Costa M, Eikmeyer F, Blom J, Prescott JF. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS One. 2012;7:e49753.
4. Sayeed S, Li J, McClane BA. Virulence plasmid diversity in Clostridium perfringens type D isolates. Infect Immun. 2007;75:2391–8.
5. Bueschel DM, Jost BH, Billington SJ, Trinh HT, Songer JG. Prevalence of cpb2, encoding beta2 toxin, in Clostridium perfringens field isolates: correlation of genotype with phenotype. Vet Microbiol. 2003;94:121–9.
6. Garmory HS, Chanter N, French NP, Bueschel D, Songer JG, Titball RW. Occurrence of Clostridium perfringens beta2-toxin amongst animals, determined using genotyping and subtyping PCR assays. Epidemiol Infect. 2000;124:61–7.
7. Bannam TL, Yan X-X, Harrison PF, Seemann T, Keyburn AL, Stubenrauch C, et al. Necrotic enteritis-derived Clostridium perfringens strain with three closely related independently conjugative toxin and antibiotic resistance plasmids. MBio. 2011;2:e00190-11.
8. Adams V, Li J, Wisniewski JA, Uzal FA, Moore RJ, McClane BA, et al. Virulence plasmids of spore-forming bacteria. Microbiol Spectr. 2014;2:PLAS-0024- 2014.
9. Freedman JC, Theoret JR, Wisniewski JA, Uzal FA, Rood JI, McClane BA. Clostridium perfringens type A–E toxin plasmids. Res Microbiol. 2015;166: 264–79.
10. Keyburn AL, Boyce JD, Vaz P, Bannam TL, Ford ME, Parker D, et al. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 2008;4:e26.
11. Nowell VJ, Kropinski AM, Songer JG, MacInnes JI, Parreira VR, Prescott JF. Genome sequencing and analysis of a type a Clostridium perfringens isolate from a case of bovine clostridial abomasitis. PLoS One. 2012;7:e32271.
12. Wong YM, Juan JC, Gan HM, Austin CM. Draft genome sequence of Clostridium perfringens strain JJC, a highly efficient hydrogen producer isolated from landfill leachate sludge. Genome Announc. 2014;2:e00064–14.
13. Johansson A, Aspan A, Bagge E, Båverud V, Engström BE, Johansson K-E. Genetic diversity of Clostridium perfringens type a isolates from animals, food poisoning outbreaks and sludge. BMC Microbiol. 2006;6:47.
14. Myers GSA, Rasko DA, Cheung JK, Ravel J, Seshadri R, DeBoy RT, et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006;16:1031–40.
15. Chalmers G, Bruce HL, Hunter DB, Parreira VR, Kulkarni RR, Jiang Y-F, et al. Multilocus sequence typing analysis of Clostridium perfringens isolates from necrotic enteritis outbreaks in broiler chicken populations. J Clin Microbiol. 2008;46:3957–64.
16. Chalmers G, Martin SW, Hunter DB, Prescott JF, Weber LJ, Boerlin P. Genetic diversity of Clostridium perfringens isolated from healthy broiler chickens at a commercial farm. Vet Microbiol. 2008;127:116–27.
17. Nauerby B, Pedersen K, Madsen M. Analysis by pulsed-field gel electrophoresis of the genetic diversity among Clostridium perfringens isolates from chickens. Vet Microbiol. 2003;94:257–66.
18. Hassan KA, Elbourne LDH, Tetu SG, Melville SB, Rood JI, Paulsen IT. Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res Microbiol. 2015;166:255–63.
19. Parish WE. Necrotic enteritis in the fowl (Gallus Gallus Domesticus). I. Histopathology of the disease and isolation of a strain of Clostridium welchii. J Comp Pathol Ther. 1961;71:377–93.
20. Songer JG. Clostridial enteric diseases of domestic animals. Clin Microbiol Rev. 1996;9:216–34.
21. Lovland A, Kaldhusdal M. Severely impaired production performance in broiler flocks with high incidence of Clostridium perfringens-associated hepatitis. Avian Pathol. 2001;30:73–81.
22. Lepp D, Roxas B, Parreira VR, Marri PR, Rosey EL, Gong J, et al. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS One. 2010;5:e10795.
23. Lepp D, Gong J, Songer JG, Boerlin P, Parreira VR, Prescott JF. Identification of accessory genome regions in poultry Clostridium perfringens isolates carrying the netB plasmid. J Bacteriol. 2013;195:1152–66.
24. Keyburn AL, Bannam TL, Moore RJ, Rood JI. NetB, a pore-forming toxin from necrotic enteritis strains of Clostridium perfringens. Toxins. 2010;2:1913–27.
25. Keyburn AL, Yan X-X, Bannam TL, Van Immerseel F, Rood JI, Moore RJ. Association between avian necrotic enteritis and Clostridium perfringens strains expressing NetB toxin. Vet Res. 2010;41:21.
26. Coursodon CF, Glock RD, Moore KL, Cooper KK, Songer JG. TpeL-producing strains of Clostridium perfringens type a are highly virulent for broiler chicks. Anaerobe. 2012;18:117–21.
27. Allaart JG, de BND, van AAJAM, Fabri THF, Gröne A. NetB-producing and beta2-producing Clostridium perfringens associated with subclinical necrotic enteritis in laying hens in the Netherlands. Avian Pathol. 2012;41(6):541.
28. Han X, Du X-D, Southey L, Bulach DM, Seemann T, Yan X-X, et al. Functional analysis of a bacitracin resistance determinant located on ICECp1, a novel Tn916-like element from a conjugative plasmid in Clostridium perfringens. Antimicrob Agents Chemother. 2015;59:6855–65.
29. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma. 2010;26:2460–1.
30. Wade B, Keyburn A, Haring V, Ford M, Rood J, Moore R. The adherent abilities of Clostridium perfringens strains are critical for the pathogenesis of avian necrotic enteritis. Vet Microbiol. 2016;197:53–61.
31. Wade B, Keyburn AL, Seemann T, Rood JI, Moore RJ. Binding of Clostridium perfringens to collagen correlates with the ability to cause necrotic enteritis in chickens. Vet Microbiol. 2015;180:299–303.
32. Timbermont L, De Smet L, Van Nieuwerburgh F, Parreira VR, Van Driessche G, Haesebrouck F, et al. Perfrin, a novel bacteriocin associated with netB positive Clostridium perfringens strains from broilers with necrotic enteritis. Vet Res. 2014;45:40.
33. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and postanalysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
34. Neumann AP, Rehberger TG. MLST analysis reveals a highly conserved core genome among poultry isolates of Clostridium septicum. Anaerobe. 2009;15: 99–106.
35. Gholamiandekhordi AR, Ducatelle R, Heyndrickx M, Haesebrouck F, Van Immerseel F. Molecular and phenotypical characterization of Clostridium perfringens isolates from poultry flocks with different disease status. Vet Microbiol. 2006;113:143–52.
36. Hughes ML, Poon R, Adams V, Sayeed S, Saputo J, Uzal FA, et al. Epsilontoxin plasmids of Clostridium perfringens type D are conjugative. J Bacteriol. 2007;189:7531–8.
37. Brynestad S, Sarker MR, McClane BA, Granum PE, Rood JI. Enterotoxin plasmid from Clostridium perfringens is conjugative. Infect Immun. 2001;69:3483–7.
38. Crespo R, Fisher DJ, Shivaprasad HL, Fernández-Miyakawa ME, Uzal FA. Toxinotypes of Clostridium perfringens isolated from sick and healthy avian species. J Vet Diagn Investig. 2007;19:329–33.
39. Brady J, Hernandez-Doria JD, Bennett C, Guenter W, House JD, RodriguezLecompte JC. Toxinotyping of necrotic enteritis-producing and commensal isolates of Clostridium perfringens from chickens fed organic diets. Avian Pathol. 2010;39:475–81.
40. Park JY, Kim S, Oh JY, Kim HR, Jang I, Lee HS, et al. Characterization of Clostridium perfringens isolates obtained from 2010 to 2012 from chickens with necrotic enteritis in Korea. Poult Sci. 2015;94:1158–64.
41. Engström BE, Johansson A, Aspan A, Kaldhusdal M. Genetic relatedness and netB prevalence among environmental Clostridium perfringens strains associated with a broiler flock affected by mild necrotic enteritis. Vet Microbiol. 2012;159:260–4.
42. Martin TG, Smyth JA. The ability of disease and non-disease producing strains of Clostridium perfringens from chickens to adhere to extracellular matrix molecules and Caco-2 cells. Anaerobe. 2010;16:533–9.
43. Barbara AJ, Trinh HT, Glock RD, Glenn Songer J. Necrotic enteritis-producing strains of Clostridium perfringens displace non-necrotic enteritis strains from the gut of chicks. Vet Microbiol. 2008;126:377–82.
44. Ronco T, Stegger M, Ng KL, Lilje B, Lyhs U, Andersen PS, et al. Genome analysis of Clostridium perfringens isolates from healthy and necrotic enteritis infected chickens and turkeys. BMC Res Notes. 2017;10:270.
45. Yother J. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol. 2011;65:563–81.
46. Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006;2:e31.
47. Seo N, Tamai K, Nishida S. Criteria for identification of Clostridium perfringens. 5. Capsule formation and urease production. Igaku To Seibutsugaku. 1969;79:215–8.
48. Baine H, Cherniak R. Capsular polysaccharides of Clostridium perfringens Hobbs 5. Biochemistry (Mosc). 1971;10:2949–52.
49. Cherniak R, Frederick HM. Capsular polysaccharide of Clostridium perfringens Hobbs 9. Infect Immun. 1977;15:765–71.
50. Sheng S, Cherniak R. Structure of the capsular polysaccharide of Clostridium perfringens Hobbs 10 determined by NMR spectroscopy. Carbohydr Res. 1997;305:65–72.
51. Stringer MF, Turnbull PC, Hughes JA, Hobbs BC. An international serotyping system for Clostridium perfringens (welchii) type a in the near future. Dev Biol Stand. 1976;32:85–9.
52. Vinogradov E, Aubry A, Logan SM. Structural characterization of wall and lipidated polysaccharides from Clostridium perfringens ATCC 13124. Carbohydr Res. 2017;448:88–94.
53. Henderson DW. The somatic antigens of the Cl. welchii group of organisms. J Hyg. 1940;40:501–12.
54. Chaguza C, Andam CP, Harris SR, Cornick JE, Yang M, Bricio-Moreno L, et al. Recombination in Streptococcus pneumoniae lineages increase with carriage duration and size of the polysaccharide capsule. MBio. 2016;7:e01053–16.
55. O’Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17:218–34.
56. Caimano MJ, Hardy GG, Yother J. Capsule genetics in Streptococcus pneumoniae and a possible role for transposition in the generation of the type 3 locus. Microb Drug Resist. 1998;4:11–23.
57. Smith HE, Damman M, van der Velde J, Wagenaar F, Wisselink HJ, StockhofeZurwieden N, et al. Identification and characterization of the cps locus of Streptococcus suis serotype 2: the capsule protects against phagocytosis and is an important virulence factor. Infect Immun. 1999;67:1750–6.
58. Prescott JF, Parreira VR, Mehdizadeh Gohari I, Lepp D, Gong J. The pathogenesis of necrotic enteritis in chickens: what we know and what we need to know: a review. Avian Pathol. 2016;45:288–94.
59. Timbermont L, Lanckriet A, Pasmans F, Haesebrouck F, Ducatelle R, Van Immerseel F. Intra-species growth-inhibition by Clostridium perfringens is a possible virulence trait in necrotic enteritis in broilers. Vet Microbiol. 2009; 137:388–91.
60. Simmons M, Donovan DM, Siragusa GR, Seal BS. Recombinant expression of two bacteriophage proteins that lyse Clostridium perfringens and share identical sequences in the C-terminal cell wall binding domain of the molecules but are dissimilar in their N-terminal active domains. J Agric Food Chem. 2010;58:10330–7.
61. Boyd EF, Brüssow H. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 2002;10:521–9.
62. Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–27.
63. Smoot LM, McCormick JK, Smoot JC, Hoe NP, Strickland I, Cole RL, et al. Characterization of two novel pyrogenic toxin superantigens made by an acute rheumatic fever clone of Streptococcus pyogenes associated with multiple disease outbreaks. Infect Immun. 2002;70:7095–104.
64. Kim K-P, Born Y, Lurz R, Eichenseher F, Zimmer M, Loessner MJ, et al. Inducible Clostridium perfringens bacteriophages ΦS9 and ΦS63. Bacteriophage. 2012;2:89–97.
65. Zimmer M, Scherer S, Loessner MJ. Genomic analysis of Clostridium perfringens bacteriophage φ3626, which integrates into guaA and possibly affects sporulation. J Bacteriol. 2002;184:4359–68.
66. Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesheater. Proc Natl Acad Sci. 2002;99:996–1001.
67. Lucchini S, Desiere F, Brüssow H. Similarly organized lysogeny modules in temperate Siphoviridae from low GC content gram-positive bacteria. Virology. 1999;263:427–35.
68. Gervasi T, Curto RL, Narbad A, Mayer MJ. Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch Virol. 2013;158:2015–7.
69. Morales CA, Oakley BB, Garrish JK, Siragusa GR, Ard MB, Seal BS. Complete genome sequence of the podoviral bacteriophage ΦCP24R, which is virulent for Clostridium perfringens. Arch Virol. 2012;157:769–72.
70. Ronco T, Stegger M, Ng KL, Lilje B, Lyhs U, Andersen PS, Pedersen K. Genome analysis of Clostridium perfringens isolates from healthy and necrotic enteritis infected chickens and turkeys. BMC Res Notes. 2017;10:270.
71. Karasov WH, Douglas AE. Comparative digestive physiology. Compr Physiol. 2013;3:741–83.
72. Aronesty E. Comparison of sequencing utlitity programs. Open Bioinform J. 2013;7:1–8.
73. Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinforma. 2012;28:1420–8.
74. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Canu PAM. Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36.
75. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963.
76. Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524.
77. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinforma. 2014; 30:2068 –9.
78. Sayers E. Entrez Programming Utilities Help. 2010 [cited 2015 Nov 18]; Available from: http://www.ncbi.nlm.nih.gov/books/NBK25501/. Accessed 18 Nov 2015 to 18 May 2018.
79. Chaudhari NM, Gupta VK, Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373.
80. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347 –52.
81. Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, et al. A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet. 2015;11:e1005413.
82. Sheedy SA, Ingham AB, Rood JI, Moore RJ. Highly conserved alpha-toxin sequences of avian isolates of Clostridium perfringens. J Clin Microbiol. 2004; 42:1345 –7.
83. Mehdizadeh Gohari I, Kropinski AM, Weese SJ, Parreira VR, Whitehead AE, Boerlin P, et al. Plasmid characterization and chromosome analysis of two netF+ Clostridium perfringens isolates associated with foal and canine necrotizing enteritis. PLoS One. 2016;11:e0148344.
84. Stanley D, Keyburn AL, Denman SE, Moore RJ. Changes in the caecal microflora of chickens following Clostridium perfringens challenge to induce necrotic enteritis. Vet Microbiol. 2012;159:155 –62.



Dr.Goodmorning, your R&D work is very use for poultry field.i need primer design and programme setting for PCR. My mail id balasubramaniank1976@gmail.com.







.jpg&w=3840&q=75)