Introduction
Significant economic losses in the poultry industries due to moderate to high mortality and decreased egg production have resulted from H9N2 low pathogenic avian influenza virus (LPAIV) infections across North Africa, the Middle East and Asia (Fusaro and others 2011, Lee and Song 2013). The currently circulating Eurasian H9N2 LPAIV has rapidly spread to become the most prevalent LPAIV in domestic poultry since their initial isolation in China during 1994 (Zhang and others 2009). Phylogenetic and antigenic analyses have identified several groups of H9N2 LPAIV in Eurasia: the G1 lineage, the Y280 lineage and the Y439/Korean lineage (Guan and others 1999, Matrosovich and others 2001, Butt and others 2010). This group of viruses has caused sporadic infections in mammalian species, including human beings, and have been associated with some specific genetic changes that suggests increasing pandemic potential (Lin and others 2000, Matrosovich and others 2001, Butt and others 2005, Cong and others 2008).
The first outbreak of the H9N2 LPAIV in Pakistan occurred in 1998 and the viruses from the outbreak were genetically close to the G1-lineage virus circulating in Hong Kong during 1997 (Cameron and others 2000). Since then, the H9N2 viruses have become prevalent in Pakistan and have evolved through reassortment with H5N1 and H7N3 highly pathogenic avian influenza viruses (HPAIVs), generating a new H9N2 genotype, represented by A/Chicken/ Pakistan/UDL-01/06 (H9N2), which has internal gene segments from G1-like H9N2, clade 2.2 H5N1 and H7N3 HPAIV (Iqbal and others 2009). Molecular characterization of H9N2 viruses collected during December 2009–February 2010 from chickens in live poultry retail shops in Lahore, Pakistan, showed that H9N2 isolates were reassortants between the G1 lineage and the H7N3 HPAIV that circulated in Pakistan and contained several mammalian host-specific markers (Chaudhry and others 2015).
Because these H9N2 novel genotypes contain mammalian host-specific markers, recent surveillance is essential to better understand any continuing public health risk. No information and genetic sequence have been reported since 2010. Here the authors report on complete genomes of new H9N2 LPAIV, three from 2015 and one from 2012, isolated from Pakistan and genetically characterised.
Materials and methods
Viruses
The authors sequenced the genome of Newcastle disease viruses collected from mortality events in Pakistan using next-generation sequencing and detected H9N2 LPAIV. Swab samples were shipped to biosafety level 3–agriculture facilities at the Southeast Poultry Research Laboratory, Agricultural Research Service, United States Department of Agriculture as frozen samples following international shipping regulations. Sample collection date and regions are shown in Fig 1. A/chicken/ Pakistan/26A/2012(H9N2), A/chicken/Pakistan/10A/ 2015(H9N2), A/chicken/Pakistan/13A/2015(H9N2) and A/pigeon/Pakistan/25A/2015(H9N2) hereafter will be designated as 26A/2012, 10A/2015, 13A/2015 and 25A/ 2015, respectively. The 10A/2015 and 26A/2012 samples were collected from a broiler chicken farm. The 13A/2015 sample was collected from a layer farm. The 25A/2015 sample was collected from a four-month-old to a five-month-old pigeon that was purchased. These viruses were propagated in nine-day-old specific pathogen free (SPF) embryonating chicken eggs.
Sequencing
Viral RNA was extracted from the allantoic fluid, using the QIAamp RNA Viral Mini Kit (Qiagen, USA). Complementary DNA was synthesised by reverse transcription reaction using Invitrogen Moloney Murine Leukemia Virus Reverse Transcriptase kit (Thermo Scientific, USA). The paired-end sequencing libraries were generated using the Nextera XT DNA Sample Preparation Kit (Illumina, USA) according to the manufacturer’s instructions. Input DNA were fragmented and tagged with sequencing adapters by Nextera XT transposome. The tagmented DNA were amplified by a reduced 12-cycle PCR programme to add index sequences required for subsequent cluster formation. The DNA libraries concentrations were measured using a Qubit 2.0 Fluorometer and the size of the libraries were determined on Bioanalyzer 2100 (Agilent Technologies, Germany) using the Agilent High Sensitivity DNA Kit (Agilent Technologies, Germany). Whole-genome sequencing (500 cycles) was conducted using Miseq benchtop next-generation sequencer (Illumina, USA). After the sequencing run, de novo and directed assembly of genome sequences were carried out using the Geneious R8 programme. The deduced amino acid sequences of polymerase basic 2 (PB2), PB1, PB1-F2, polymerase acidic (PA), PA-X, haemagglutinin (HA), nucleocapsid (NP), neuraminidase (NA), matrix 1, matrix 2, non-structural 1 (NS1) and NS2 proteins were analysed using the Geneious R8 programme. Nucleotide sequences of four H9N2 LPAIVs have been deposited in GenBank under accession no. KU042891-KU042922.
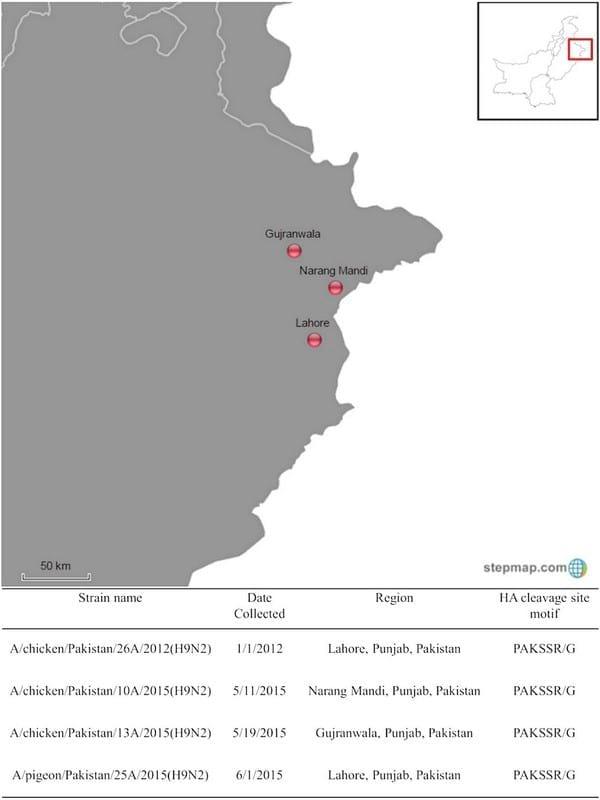
FIG 1: Virus used in this study. Red circle buttons identify the location of identified viruses
Phylogenetic analysis
The nucleotide sequences of HA segment were aligned using MUSCLE (Edgar 2004). The Bayesian relaxed clock phylogenetic analyses were done using BEAST V.1.8.2 (Drummond and Rambaut 2007). The authors applied an uncorrelated lognormal distribution relaxed clock method, the SRD06 nucleotide substitution model and the Bayesian skyline coalescent prior. A Markov Chain Monte Carlo method to sample trees and evolutionary parameters was run for 5.0×107 generations. At least three independent chains were combined to ensure adequate sampling of the posterior distribution of trees. BEAST output was analysed with TRACER V.1.4 with 10 per cent burn-in. The FigTree V.1.4.2 programme was used to construct and visualise the maximum clade credibility tree. Maximum-likelihood phylogenies of PB2, PB1, PA, NP, NA, M, non-structural (NS) segments were generated with RAxML using the general time reversible nucleotide substitution model, with among-site rate variation modelled using a discrete γ distribution. Bootstrap support values were generated using 1000 rapid bootstrap replicates and represented on nodes.
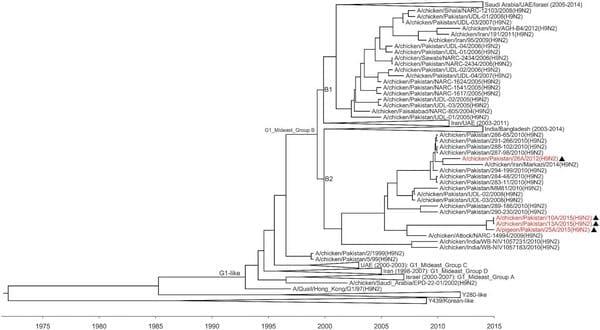
FIG 2: Relaxed clock molecular phylogenetic tree for the haemagglutinin of H9N2 viruses identified in Pakistan. The phylogenetic relationships and temporal evolutionary history have been estimated by molecular clock analysis. Black triangles indicate viruses detected and characterised in the present study
Results
All of the viruses tested in this study were LPAIV based on amino acid sequence, PAKSSR/G, at the HA proteolytic cleavage site. All segments of the viruses sequenced in this study, 26A/2012, 10A/2015, 13A/ 2015 and 25A/2015, belonged to Middle East group B (see Fig 2 and online supplementary figure) (Fusaro and others 2011). The PB1, PA, NP, M and NS segments were clustered with A/chicken/Karachi/ NARC-100/2004(H7N3) virus. As shown in Fig 2, the HA gene of H9N2 viruses identified in Pakistan from 2004 to 2015 belonged to Middle East group B and fell within two separate sublineages, designated as sublineage B1, which contains viruses collected from 2004 to 2008 and clustered with other Middle East viruses, and sublineage B2, which contains viruses collected from 2008 to 2015 and clustered with viruses identified from the Indian subcontinent.
The authors analysed the deduced amino acid sequences of viral proteins to identify the possible determinants of zoonotic transmission (Table 1). All of the viruses sequenced in this study had an identical amino acid residues leucine (L) at position 226 in the receptorbinding site (RBS) of HA gene. Based on the deduced amino acid sequence of the internal genes, 10A/2015, 13A/2015 and 25A/2015 viruses also contained other mammalian host-specific markers (Chaudhry and others 2015) in the matrix 1 protein (V15I); the C-terminus of PDZ ligand motif in NS1 protein (KSEI); and the NP (E372D) protein. The 26A/2012 virus contained mammalian host-specific markers in the M2 protein (L55F); the C-terminus of PDZ ligand motif in NS1 protein (KSEI); the NP protein (E372D) protein.
Discussion
The H9N2 LPAIV isolated from 1998 to 2010 in Central Asia and the Middle East comprise four distinct groups (A, B, C and D) with groups A and B circulating extensively in Central Asia and the Middle East since 1999 (Fusaro and others 2011). As shown in Fig 1, H9N2 viruses identified in Pakistan from 2012 and 2015 belonged to sublineage B1, which contains viruses collected since 2008 and clustered with viruses identified from the Indian subcontinent. The presence of lysine at position 4 in the HA proteolytic cleavage site was observed in H9 isolates from the Indian subcontinent (Iqbal and others 2009, Shanmuganatham and others 2013, Chaudhry and others 2015). All of the viruses tested in this study had lysine at position 4 in the HA proteolytic cleavage site.
The H9N2 LPAIV identified in Pakistan from 2006 to 2010 were reassortants between the G1 lineage and the H7N3 HPAIV that circulated in Pakistan and belong to genetic group B (Fusaro and others 2011, Chaudhry and others 2015). Molecular characterisation of H9N2 viruses collected during December 2009–February 2010 from chickens in live poultry retail shops in Lahore, Pakistan, showed that PB1, PA and NS genes of these viruses originated by reassortment events from local H7N3 HPAIVs circulating in Pakistan (Chaudhry and others 2015). All segments of the viruses sequenced in this study were clustered with these 2009–2010 viruses. However, in the present phylogenetic study, NP and M genes of A/chicken/Karachi/NARC-100/2004(H7N3) virus were also clustered with H9N2 viruses identified from Middle East, Pakistan and Bangladesh. Consistent with the present phylogenetic analysis, the PB1, PA, NP, M and NS segments of H9N2 viruses identified in Bangladesh between 2011 and 2013 were clustered with A/chicken/Karachi/NARC-100/ 2004(H7N3) virus and H9N2 viruses identified from Pakistan (Shanmuganatham and others 2014).
The G1 lineage H9N2 LPAIV has been sporadically isolated from human beings suffering from flu-like illness (Lin and others 2000), and are predicted to have an affinity for the human receptor binding profile (Matrosovich and others 2001). The amino acid L at position 226 in the HA RBS plays a key role in human virus-like receptor specificity and promotes the transmission of H9N2 in ferrets (Wan and others 2008). Sequence comparison showed that the 2009–2010 Pakistani H9N2 viruses (Chaudhry and others 2015) and all of the viruses sequenced in this study had an identical amino acid residue L at position 226 in the HA RBS. As shown in Table 1, the mammalian host-specific markers found in this study suggest that Pakistani H9N2 viruses are still potentially infectious for mammals. The sequence analyses of internal genes showed that these viruses also contained mammalian host-specific markers, including the aspartic acid at position 372 in NP protein, the isoleucine at position 15 in M1 protein, the phenylalanine at position 55 in M2 protein and uncommon PDZ ligand motif. The aspartic acid at position 372 in NP protein has been found in avian influenza viruses isolated from human beings (Chen and others 2006, Iqbal and others 2009, Pan and others 2010). The isoleucine at position 15 in M1 protein was associated with high pathogenicity of H5N1 viruses in mice (Katz and others 2000, Iqbal and others 2009). The phenylalanine at position 55 in M2 protein is a putative molecular determinant responsible for swine-origin influenza A virus transmission in human beings (Pan and Jiang 2009). Viruses containing PDZ ligand motif from the 1918 H1N1 and H5N1 HPAIV demonstrated increased virulence in infected mice (Jackson and others 2008). All viruses contained an uncommon KSEI sequence as a PDZ ligand motif in the NS protein.
Considering the possibility of future gene reassortment with additional HPAIV and the presence of what appear to be mammalian host-specific markers, the public health threat of H9N2 viruses in Pakistan and other countries continues. Continued active surveillance in poultry and mammals is needed to monitor the spread and understand the potential for zoonotic infection.
Correction notice The author name Claudio L. Afonso has been corrected.
Acknowledgements The authors thank Dawn Williams-Coplin and Tim Olivier for excellent technical assistance.
References
Butt A. M., Siddique S., Idrees M., Tong Y. (2010) Avian influenza A (H9N2): computational molecular analysis and phylogenetic characterization of viral surface proteins isolated between 1997 and 2009 from the human population. Virology Journal 7, 319
Butt K. M., Smith G. J., Chen H., Zhang L. J., Leung Y. H., Xu K. M., Lim W., Webster R. G., Yuen K. Y., Peiris J. S., Guan Y. (2005) Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. Journal of Clinical Microbiology 43, 5760–5767
Cameron K. R., Gregory V., Banks J., Brown I. H., Alexander D. J., Hay A. J., Lin Y. P. (2000) H9N2 subtype influenza A viruses in poultry in pakistan are closely related to the H9N2 viruses responsible for human infection in Hong Kong. Virology 278, 36–41
Chaudhry M., Angot A., Rashid H. B., Cattoli G., Hussain M., Trovò G., Drago A., Valastro V., Thrusfield M., Welburn S., Eisler M. C., Capua I. (2015) Reassortant Avian Influenza A(H9N2) viruses in chickens in retail poultry shops, Pakistan, 2009–2010. Emerging Infectious Diseases 21, 673–676
Chen G. W., Chang S. C., Mok C. K., Lo Y. L., Kung Y. N., Huang J. H., Shih Y. H., Wang J. Y., Chiang C., Chen C. J., Shih S. R. (2006) Genomic signatures of human versus avian influenza A viruses. Emerging Infectious Diseases 12, 1353–1360
Cong Y. L., Wang C. F., Yan C. M., Peng J. S., Jiang Z. L., Liu J. H. (2008) Swine infection with H9N2 influenza viruses in China in 2004. Virus Genes 36, 461–469
Drummond A. J., Rambaut A. (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7, 214 Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797
Fusaro A., Monne I., Salviato A., Valastro V., Schivo A., Amarin N. M., Gonzalez C., Ismail M. M., Al-Ankari A. R., Al-Blowi M. H., Khan O. A., Maken Ali A. S., Hedayati A., Garcia Garcia J., Ziay G. M., Shoushtari A., Al Qahtani K. N., Capua I., Holmes E. C., Cattoli G. (2011) Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications. Journal of Virology 85, 8413–8421
Guan Y., Shortridge K. F., Krauss S., Webster R. G. (1999) Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal" genes of H5N1 viruses in Hong Kong? Proceedings of the National Academy of Sciences of the United States of America 96, 9363–9367
Iqbal M., Yaqub T., Reddy K., Mccauley J. W. (2009) Novel genotypes of H9N2 influenza A viruses isolated from poultry in Pakistan containing NS genes similar to highly pathogenic H7N3 and H5N1 viruses. PLoS ONE 4, e5788
Jackson D., Hossain M. J., Hickman D., Perez D. R., Lamb R. A. (2008) A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proceedings of the National Academy of Sciences of the United States of America 105, 4381–4386
Katz J. M., Lu X., Tumpey T. M., Smith C. B., Shaw M. W., Subbarao K. (2000) Molecular correlates of influenza A H5N1 virus pathogenesis in mice. Journal of Virology 74, 10807–10810
Lee D. H., Song C. S. (2013) H9N2 avian influenza virus in Korea: evolution and vaccination. Clinical and Experimental Vaccine Research 2, 26–33
Lin Y. P., Shaw M., Gregory V., Cameron K., Lim W., Klimov A., Subbarao K., Guan Y., Krauss S., Shortridge K., Webster R., Cox N., Hay A. (2000) Avian-to-human transmission of H9N2 subtype influenza A viruses: relationship between H9N2 and H5N1 human isolates. Proceedings of the National Academy of Sciences of the United States of America 97, 9654–9658
Matrosovich M. N., Krauss S., Webster R. G. (2001) H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Archives of Virology 281, 156–162
Pan C., Cheung B., Tan S., Li C., Li L., Liu S., Jiang S. (2010) Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus. PLoS ONE 5, e9549
Pan C., Jiang S. (2009) E14-F55 combination in M2 protein: a putative molecular determinant responsible for swine-origin influenza A virus transmission in humans. PLoS Currents Influenza 1, RRN1044 Shanmuganatham K., Feeroz M. M., Jones-Engel L., Smith G. J., Fourment M., Walker D., Mcclenaghan L., Alam S. M., Hasan M. K., Seiler P., FranksJ., Danner A., Barman S., Mckenzie P., Krauss S., Webby R. J., Webster R. G. (2013) Antigenic and molecular characterization of avian influenza A(H9N2) viruses, Bangladesh. Emerging Infectious Diseases 19
Shanmuganatham K., Feeroz M. M., Jones-Engel L., Walker D., Alam S., Hasan M., Mckenzie P., Krauss S., Webby R. J., Webster R. G. (2014) Genesis of avian influenza H9N2 in Bangladesh. Emerging Infectious Diseases 3, e88
Wan H., Sorrell E. M., Song H., Hossain M. J., Ramirez-Nieto G., Monne I., Stevens J., Cattoli G., Capua I., Chen L. M., Donis R. O., Busch J., Paulson J. C., Brockwell C., Webby R., Blanco J., Al-Natour M. Q., Perez D. R. (2008) Replication and transmission of H9N2 influenza viruses in ferrets: evaluation of pandemic potential. PLoS ONE 3, e2923
Zhang P., Tang Y., Liu X., Liu W., Zhang X., Liu H., Peng D., Gao S., Wu Y., Zhang L., Lu S., Liu X. (2009) A novel genotype H9N2 influenza virus possessing human H5N1 internal genomes has been circulating in poultry in eastern China since 1998. Journal of Virology 83, 8428–8438











.jpg&w=3840&q=75)