Fast and Slow-Growing Management Systems: Characterisation of Broiler Caecal Microbiota Development throughout the Growing Period
Author details:
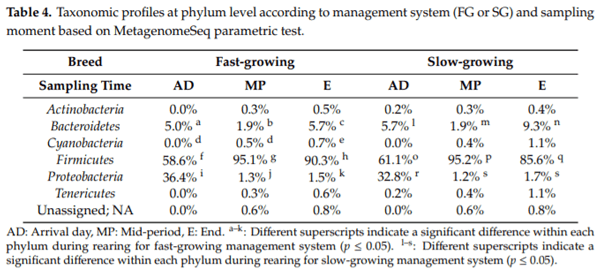
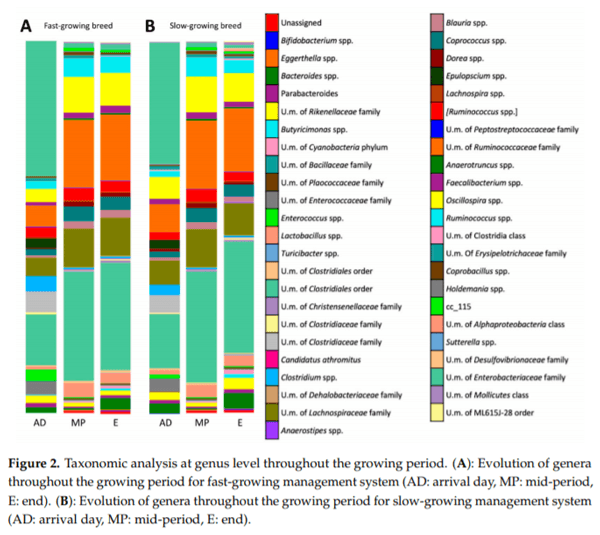
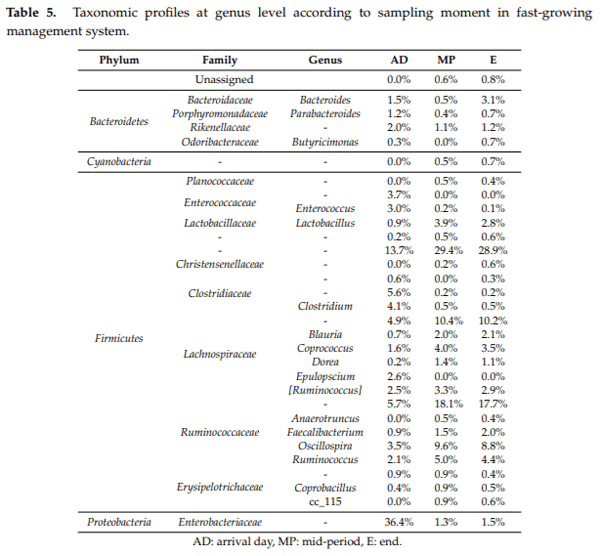
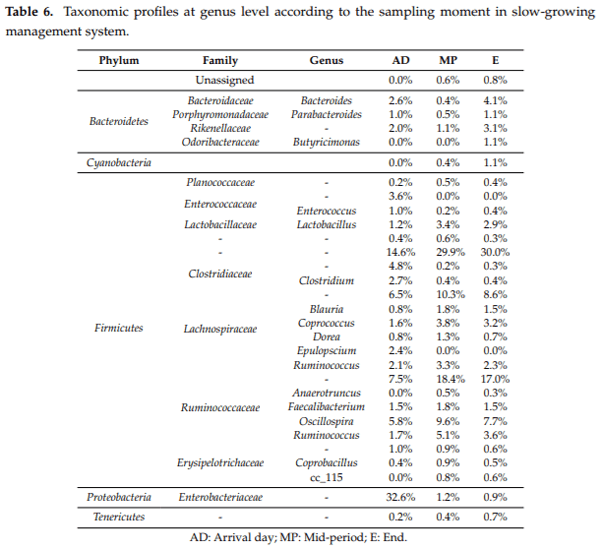
Caecal microbiota and its modulation play an important role in poultry health, productivity and disease control. Moreover, due to the emergence of antimicrobial-resistant bacteria, society is pressing for a reduction in antibiotic administration by finding effective alternatives at farm level, such as less intensified production systems. Hence, the aim of this study was to characterise the caecal microbiota in two different broiler management systems, fast and slow-growing, using 16S rRNA sequencing analysis. To this end 576 broilers were reared in two different management systems (fast and slow-growing). Results showed that Firmicutes represented the dominant phylum for both systems. At the onset, Proteobacteria was the second prevalent phylum for fast and slow-growing breeds, outnumbering the Bacteroidetes. However, during the rest of the production cycle, Bacteroidetes was more abundant than Proteobacteria in both groups. Finally, regardless of the management system, the most predominant genera identified were Oscillospira spp., Ruminococcus spp., Coprococcus spp., Lactobacillus spp. and Bacteroides spp. In conclusion, fast and slow-growing broiler microbiota are in constant development throughout rearing, being relatively stable at 21 days of age. Regarding the genus, it should be noted that the three most abundant groups for both systems, Ruminococcus spp., Lactobacillus spp. and Bacteroides spp., are related to better productive performance and intestinal health.
Keywords: broiler; growing period; microbiota; biomarker; 16S rRNA analysis.
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [CrossRef] [PubMed]
- Stanley, D.; Hughes, R.J.; Moore, R.J. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 2014, 98, 4301–4310. [CrossRef] [PubMed]
- Pourabedin, M.; Zhao, X. Prebiotics and gut microbiota in chickens. FEMS Microbiol. Lett. 2015, 362, 1–8. [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14. [CrossRef]
- Banerjee, S.; Sar, A.; Misra, A.; Pal, S.; Chakraborty, A.; Dam, B. Increased productivity in poultry birds by sub-lethal dose of antibiotics is arbitrated by selective enrichment of gut microbiota, particularly short-chain fatty acid producers. Microbiology (United Kingdom) 2018, 164, 142–153. [CrossRef]
- Clavijo, V.; Flórez, M.J.V. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: A review. Poult. Sci. 2018, 97, 1006–1021. [CrossRef]
- Pandit, R.J.; Hinsu, A.T.; Patel, N.V.; Koringa, P.G.; Jakhesara, S.J.; Thakkar, J.R.; Shah, T.M.; Limon, G.; Psifidi, A.; Guitian, J.; et al. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome 2018, 6, 1–13. [CrossRef]
- Shang, Y.; Kumar, S.; Oakley, B.; Kim, W.K. Chicken gut microbiota: Importance and detection technology. Front. Vet. Sci. 2018, 5. [CrossRef]
- Carrasco, J.M.D.; Casanova, N.A.; Miyakawa, M.E.F. Microbiota, gut health and chicken productivity: What is the connection? Microorganisms 2019, 7, 374. [CrossRef]
- Sekirov, I.; Russell, S.L.; Caetano, M.; Antunes, L.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [CrossRef]
- Kers, J.G.; Velkers, F.C.; Fischer, E.A.J.; Hermes, G.D.A.; Stegeman, J.A.; Smidt, H. Host and environmental factors a ecting the intestinal microbiota in chickens. Front. Microbiol. 2018, 9, 1–14. [CrossRef] [PubMed]
- WHO|Antimicrobial Resistance: Global Report on Surveillance 2014; World Health Organization: Geneva, Switzerland, 2014.
- Alós, J. Resistencia bacteriana a los antibióticos: Una crisis global. Enferm. Infecc. Microbiol. Clin. 2015, 33, 692–699. [CrossRef] [PubMed]
- Gadde, U.; Kim, W.H.; Oh, S.T.; Lillehoj, H.S. Alternatives to antibiotics for maximizing growth performance and feed e ciency in poultry: A review. Anim. Health Res. Rev. 2017, 18, 26–45. [CrossRef]
- Montoro-Dasi, L.; Villagra, A.; Sevilla-Navarro, S.; Pérez-Gracia, M.T.; Vega, S.; Marin, C. The dynamic of antibiotic resistance in commensal Escherichia coli throughout the growing period in broiler chickens: Fast-growing vs. slow-growing breeds. Poult. Sci. 2020, 99, 1591–1597. [CrossRef] [PubMed]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [CrossRef] [PubMed]
- Cheng, G.; Hao, H.; Xie, S.; Wang, X.; Dai, M.; Huang, L.; Yuan, Z. Antibiotic alternatives: The substitution of antibiotics in animal husbandry? Front. Microbiol. 2014, 5, 1–15. [CrossRef]
- Castellini, C.; Bosco, A.D. Animal Welfare and Poultry Meat in Alternative Production Systems (and Ethics of Poultry Meat Production); Elsevier Ltd.: Amsterdam, The Netherlands, 2017; ISBN 9780081007631.
- Polycarpo, G.V.; Andretta, I.; Kipper, M.; Dadalt, J.C.; Rodrigues, P.H.M.; Albuquerque, R. Meta-analytic study of organic acids as an alternative performance-enhancing feed additive to antibiotics for broiler chickens. Poult. Sci. 2017, 96, 3645–3653. [CrossRef]
- Suresh, G.; Das, R.K.; Brar, S.K.; Rouissi, T.; Ramirez, A.; Chorfi, Y.; Godbout, S. Critical Reviews in Microbiology Alternatives to antibiotics in poultry feed: Molecular perspectives. Crit. Rev. Microbiol. 2017, 1–18. [CrossRef]
- Alagawany, M.; Abd El-Hack, M.E.; Farag, M.R.; Sachan, S.; Karthik, K.; Dhama, K. The use of probiotics as eco-friendly alternatives for antibiotics in poultry nutrition. Environ. Sci. Pollut. Res. 2018, 25, 10611–10618. [CrossRef]
- Sevilla-Navarro, S.; Marín, C.; Cortés, V.; García, C.; Vega, S.; Catalá-Gregori, P. Autophage as a control measure for Salmonella in laying hens. Poult. Sci. 2018, 97, 4367–4373. [CrossRef]
- Kogut, M.H.; Yin, X.; Yuan, J.; Broom, L. Gut health in poultry. CAB Rev. Perspect. Agric. Vet. Sci. Nutr. Nat. Resour. 2017, 12. [CrossRef]
- Pedroso, A.A.; Menten, J.F.M.; Lambais, M.R. The Structure of Bacterial Community in the Intestines of Newly Hatched Chicks. J. Appl. Poult. Res. 2005, 14, 232–237. [CrossRef]
- Oakley, B.B.; Morales, C.A.; Line, J.; Berrang, M.E.; Meinersmann, R.J.; Tillman, G.E.; Wise, M.G.; Siragusa, G.R.; Hiett, K.L.; Seal, B.S. The Poultry-Associated Microbiome: Network Analysis and Farm-to-Fork Characterizations. PLoS ONE 2013, 8. [CrossRef] [PubMed]
- Richards, P.; Fothergill, J.; Bernardeau, M.; Wigley, P. Development of the caecal microbiota in three broiler breeds. Front. Vet. Sci. 2019, 6. [CrossRef]
- Xi, Y.; Shuling, N.; Kunyuan, T.; Qiuyang, Z.; Hewen, D.; ChenCheng, G.; Tianhe, Y.; Liancheng, L.; Xin, F. Characteristics of the intestinal flora of specific pathogen free chickens with age. Microb. Pathog. 2019, 132, 325–334. [CrossRef]
- Brisbin, J.T.; Gong, J.; Sharif, S. Interactions between commensal bacteria and the gut-associated immune system of the chicken. Anim. Health Res. Rev. 2008, 9, 101–110. [CrossRef]
- Rasschaert, G.; Houf, K.; Godard, C.; Wildemauwe, C.; Pastuszczak-Frak,? M.; De Zutter, L. Contamination of carcasses with Salmonella during poultry slaughter. J. Food Prot. 2008, 71, 146–152. [CrossRef]
- Ellerbroek, L.I.; Lienau, J.A.; Klein, G. Campylobacter spp. in broiler flocks at farm level and the potential for cross-contamination during slaughter. Zoonoses Public Health 2010, 57. [CrossRef]
- Sevilla-Navarro, S.; Marin, C.; Cortés, V.; García, C.; Catalá-Gregori, P. Campylobacter prevalence and risk factors associated with exceeding allowable limits in poultry slaughterhouses in Spain. Vet. Rec. 2020, 186. [CrossRef]
- Kogut, M.H. The e ect of microbiome modulation on the intestinal health of poultry. Anim. Feed Sci. Technol. 2019, 250, 32–40. [CrossRef]
- Spain 2013. Royal Degree 53/2013, 1st of Febrary, por el que se Establecen las Normas Básicas Aplicables Para la Protección de los Animales Utilizados en Experimentación y Otros Fines Científicos, Incluyendo la Docencia; Boletín Oficial del Estado: Madrid, Spain, 2013; Volume 34, pp. 11370–11421.
- Ross. Ross 308/Ross 308 FF broiler: Performance Objectives. Available online: http://es.aviagen.com/assets/Tech_Center/Ross_Broiler/Ross308-308FF-BroilerPO2019-EN.pdf (accessed on 17 June 2020).
- Valls, M. Reproductoras y Pollos de Crecimiento Lento Como Producto Diferenciado. Available online: https://avicultura.info/reproductoras-y-pollos-de-crecimiento-lento/ (accessed on 17 June 2020).
- Santomá, G.; Mateos, G.G. Necesidades Nutricionales para Avicultura: Normas FEDNA, 2nd ed.; FEDNA (Fundación Española Desarrollo Nutrición Animal): Madrid, Spain; ISBN 9788409065295.
- Illumina Support. 16S Metagenomic Sequencing Library Preparation. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 17 June 2020).
- Babraham Bioinformatics. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 June 2020).
- Babraham Bioinformatics. Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 17 June 2020).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; P?a, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [CrossRef] [PubMed]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16. [CrossRef] [PubMed]
- Qiime (Quantitative Insights Into Microbial Ecology). Compare_alpha_diversity.py–This Script Compares Alpha Diversities Based on a Two-Sample t-test Using Either Parametric or Non-Parametric (Monte Carlo) Methods. Available online: http://qiime.org/scripts/compare_alpha_diversity.html (accessed on 18 June 2020).
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Di erential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10. [CrossRef]
- Mancabelli, L.; Ferrario, C.; Milani, C.; Mangifesta, M.; Turroni, F.; Duranti, S.; Lugli, G.A.; Viappiani, A.; Ossiprandi, M.C.; van Sinderen, D.; et al. Insights into the biodiversity of the gut microbiota of broiler chickens. Environ. Microbiol. 2016, 18, 4727–4738. [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [CrossRef]
- Hasan, N.; Yang, H. Factors a ecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 8, 1–31. [CrossRef]
- Wang, J.; Nesengani, L.T.; Gong, Y.; Yang, Y.; Lu, W. 16S rRNA gene sequencing reveals e ects of photoperiod on cecal microbiota of broiler roosters. PeerJ 2018, 2. [CrossRef]
- Lu, J.; Idris, U.; Harmon, B.; Hofacre, C.; Maurer, J.J.; Lee, M.D. Diversity and Succession of the Intestinal Bacterial Community of the Maturing Broiler Chicken. Appl. Environ. Microbiol. 2003, 69, 6816–6824. [CrossRef]
- Mohd Shaufi, M.A.; Sieo, C.C.; Chong, C.W.; Gan, H.M.; Ho, Y.W. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015, 7, 1–12. [CrossRef]
- Ocejo, M.; Oporto, B.; Hurtado, A. 16S rRNA amplicon sequencing characterization of caecal microbiome composition of broilers and free-range slow-growing chickens throughout their productive lifespan. Sci. Rep. 2019, 9, 1–14. [CrossRef]
- Stanley, D.; Denman, S.E.; Hughes, R.J.; Geier, M.S.; Crowley, T.M.; Chen, H.; Haring, V.R.; Moore, R.J. Intestinal microbiota associated with di erential feed conversion e ciency in chickens. Appl. Microbiol. Biotechnol. 2012, 96, 1361–1369. [CrossRef] [PubMed]
- Schokker, D.; Veninga, G.; Vastenhouw, S.A.; Bossers, A.; de Bree, F.M.; Kaal-Lansbergen, L.M.T.E.; Rebel, J.M.J.; Smits, M.A. Early life microbial colonization of the gut and intestinal development di er between genetically divergent broiler lines. BMC Genom. 2015, 16. [CrossRef] [PubMed]
- Qu, A.; Brulc, J.M.; Wilson, M.K.; Law, B.F.; Theoret, J.R.; Joens, L.A.; Konkel, M.E.; Angly, F.; Dinsdale, E.A.; Edwards, R.A.; et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE 2008, 3. [CrossRef]
- Wei, S.; Morrison, M.; Yu, Z. Bacterial census of poultry intestinal microbiome. Poult. Sci. 2013, 92, 671–683. [CrossRef]
- Rychlik, I. Composition and function of chicken gut microbiota. Animals 2020, 10, 103. [CrossRef]
- Ballou, A.L.; Ali, R.A.; Mendoza, M.A.; Ellis, J.C.; Hassan, H.M.; Croom, W.J.; Koci, M.D. Development of the chick microbiome: How early exposure influences future microbial diversity. Front. Vet. Sci. 2016, 3. [CrossRef]
- Ducatelle, R.; Goossens, E.; De Meyer, F.; Eeckhaut, V.; Antonissen, G.; Haesebrouck, F.; Van Immerseel, F. Biomarkers for monitoring intestinal health in poultry: Present status and future perspectives. Vet. Res. 2018, 49, 1–9. [CrossRef]
- Yacoubi, N.; Saulnier, L.; Bonnin, E.; Devillard, E.; Eeckhaut, V.; Rhayat, L.; Ducatelle, R.; Van Immerseel, F. Short-chain arabinoxylans prepared from enzymatically treated wheat grain exert prebiotic e ects during the broiler starter period. Poult. Sci. 2018, 97, 412–424. [CrossRef]
- Kumar, S.; Chen, C.; Indugu, N.; Werlang, G.O.; Singh, M.; Kim, W.K.; Thippareddi, H. E ect of antibiotic withdrawal in feed on chicken gut microbial dynamics, immunity, growth performance and prevalence of foodborne pathogens. PLoS ONE 2018, 13, 1–23. [CrossRef]
- Neal-McKinney, J.M.; Lu, X.; Duong, T.; Larson, C.L.; Call, D.R.; Shah, D.H.; Konkel, M.E. Production of Organic Acids by Probiotic Lactobacilli Can Be Used to Reduce Pathogen Load in Poultry. PLoS ONE 2012, 7. [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [CrossRef] [PubMed]
- The European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17. [CrossRef]
- Zhao, L.; Wang, G.; Siegel, P.; He, C.; Wang, H.; Zhao, W.; Zhai, Z.; Tian, F.; Zhao, J.; Zhang, H.; et al. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci. Rep. 2013, 3. [CrossRef]
- Torok, V.A.; Hughes, R.J.; Mikkelsen, L.L.; Perez-Maldonado, R.; Balding, K.; MacAlpine, R.; Percy, N.J.; Ophel-Keller, K. Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials. Appl. Environ. Microbiol. 2011, 77, 5868–5878. [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3. [CrossRef]
- Siegerstetter, S.C.; Schmitz-Esser, S.; Magowan, E.; Wetzels, S.U.; Zebeli, Q.; Lawlor, P.G.; O’Connell, N.E.; Metzler-Zebeli, B.U. Intestinal microbiota profiles associated with low and high residual feed intake in chickens across two geographical locations. PLoS ONE 2017, 12, 1–23. [CrossRef]
- Jha, R.; Singh, A.K.; Yadav, S.; Berrocoso, J.F.D.; Mishra, B. Early Nutrition Programming (in ovo and Post-hatch Feeding) as a Strategy to Modulate Gut Health of Poultry. Front. Vet. Sci. 2019, 6. [CrossRef]
- Yadav, S.; Jha, R. Strategies to modulate the intestinal microbiota and their e ects on nutrient utilization, performance, and health of poultry. J. Anim. Sci. Biotechnol. 2019, 10, 1–11. [CrossRef]
- Zhu, X.Y.; Zhong, T.; Pandya, Y.; Joerger, R.D. 16S rRNA-based analysis of microbiota from the cecum of broiler chickens. Appl. Environ. Microbiol. 2002, 68, 124–137. [CrossRef]
- Rodríguez, M.L.; Rebolé, A.; Velasco, S.; Ortiz, L.T.; Treviño, J.; Alzueta, C. Wheat- and barley-based diets with or without additives influence broiler chicken performance, nutrient digestibility and intestinal microflora. J. Sci. Food Agric. 2012, 92, 184–190. [CrossRef]
- Paraskeuas, V.; Mountzouris, K.C. Broiler gut microbiota and expressions of gut barrier genes a ected by cereal type and phytogenic inclusion. Anim. Nutr. 2019, 5, 22–31. [CrossRef] [PubMed]
- Pan, D.; Yu, Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes 2014, 5, 108–119. [CrossRef] [PubMed]
- Chen, J.; Tellez, G.; Richards, J.D.; Escobar, J. Identification of potential biomarkers for gut barrier failure in broiler chickens. Front. Vet. Sci. 2015, 2. [CrossRef] [PubMed]
- Fernandes, J.I.M.; Lima, F.R.; Mendonça, C.X.; Mabe, I.; Albuquerque, R.; Leal, P.M. Relative bioavailability of phosphorus in feed and agricultural phosphates for poultry. Poult. Sci. 1999, 78, 1729–1736. [CrossRef]








.jpg&w=3840&q=75)